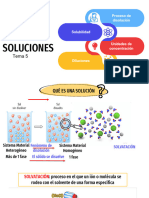
Clase11 Soluciones
Clase11 Soluciones
Descargar como pdf o txt
También podría gustarte
- INTRODUCCION Organica HidrocarburosDocumento48 páginasINTRODUCCION Organica Hidrocarburosjhamer rivera nuñezAún no hay calificaciones
- Presentación Soluciones Parte 1Documento16 páginasPresentación Soluciones Parte 1patotapia403893Aún no hay calificaciones
- SolucionesDocumento92 páginasSolucionesValentina EstupiñanAún no hay calificaciones
- Tema4 SOLUCIONESDocumento53 páginasTema4 SOLUCIONESValenkawaii ZerpaAún no hay calificaciones
- SESION 8-Propiedades de Los Líquidos.Documento25 páginasSESION 8-Propiedades de Los Líquidos.Gabriel Armando Orellana NolascoAún no hay calificaciones
- Clase Teórico 06.08.20Documento41 páginasClase Teórico 06.08.20Eze AusterlitzAún no hay calificaciones
- ACFrOgAjAmmYsoZf8k8J7lEs - w9FxVHTeGB2X-s71O-PBtbi7bNL4Wy3aRjPwKwooZMvbyZz21t SgLS3 O8BwxC1O9KnMZqQ0efLvBupH1NJJKc6EAZmvytGsl4wJy0qIJxP5gna27XgQVQjN4Documento39 páginasACFrOgAjAmmYsoZf8k8J7lEs - w9FxVHTeGB2X-s71O-PBtbi7bNL4Wy3aRjPwKwooZMvbyZz21t SgLS3 O8BwxC1O9KnMZqQ0efLvBupH1NJJKc6EAZmvytGsl4wJy0qIJxP5gna27XgQVQjN4Alfaro Barrientos Diego MaximilianoAún no hay calificaciones
- Unidad 5 - SOLUCIONES PDFDocumento23 páginasUnidad 5 - SOLUCIONES PDFGiuttaroAún no hay calificaciones
- Tema8 DisolucionesDocumento38 páginasTema8 DisolucionesCarmelo Rodríguez RomeroAún no hay calificaciones
- DisolucionesDocumento33 páginasDisolucionesNicolas EHAún no hay calificaciones
- Unidad VI SolucionesDocumento22 páginasUnidad VI SolucionesMiaAún no hay calificaciones
- Archivodiapositiva 202211292275Documento26 páginasArchivodiapositiva 202211292275JHON JAIRO YEPEZ ROJASAún no hay calificaciones
- SOLUCIONESDocumento7 páginasSOLUCIONESLino AndresAún no hay calificaciones
- CLASE 9 - DisolucionesDocumento43 páginasCLASE 9 - DisolucionesMarcelo VissaniAún no hay calificaciones
- SolucionesDocumento48 páginasSolucionesDavid AriasAún no hay calificaciones
- Unidad 5 - SOLUCIONESDocumento16 páginasUnidad 5 - SOLUCIONESMiguel Gomez MacielAún no hay calificaciones
- Parte I Soluciones 1Documento51 páginasParte I Soluciones 1Luis LópezAún no hay calificaciones
- Clase de Soluciones CompletaDocumento108 páginasClase de Soluciones CompletaAlison EscobarAún no hay calificaciones
- Unidad V Parte B - Soluciones IIDocumento21 páginasUnidad V Parte B - Soluciones IISergio MontesAún no hay calificaciones
- IntroDocumento58 páginasIntroJean QcAún no hay calificaciones
- Presentacion Soluciones QuimicaDocumento36 páginasPresentacion Soluciones QuimicaMariana SotoAún no hay calificaciones
- Unidad N º 3Documento19 páginasUnidad N º 3Jesus Calderon Zambrana100% (1)
- Apuntes de SolucionesDocumento11 páginasApuntes de SolucionesRoberto SantiagoAún no hay calificaciones
- So Luci OnesDocumento42 páginasSo Luci OnesEsteban RodriguezAún no hay calificaciones
- SolucionesDocumento28 páginasSolucionesSergio GaonaAún no hay calificaciones
- 4 SolucionesDocumento13 páginas4 Solucioneswacom76096Aún no hay calificaciones
- SolucionesDocumento7 páginasSolucionesArturo MurguiaAún no hay calificaciones
- Clase Unidades de ConcentraciónDocumento16 páginasClase Unidades de Concentraciónm.mbiggioAún no hay calificaciones
- SolucionesDocumento92 páginasSolucionesNicolas EH100% (1)
- Trabajo de Química, Soluciones VistaDocumento20 páginasTrabajo de Química, Soluciones VistaLuis Matos63% (8)
- Quimica General SolucionesDocumento22 páginasQuimica General SolucionesFederico David FloresAún no hay calificaciones
- SOLUBILIDADDocumento55 páginasSOLUBILIDADAnahis EricesAún no hay calificaciones
- Unidad 7 SolucionesDocumento157 páginasUnidad 7 SolucionesMilagros Min MoralesAún no hay calificaciones
- SOLUCIONES 2022 - Profesor GAVANCHODocumento30 páginasSOLUCIONES 2022 - Profesor GAVANCHODiego Sebastian Pumayalli CaceresAún no hay calificaciones
- Tema 2 - Las DisolucionesDocumento36 páginasTema 2 - Las Disolucionesstefanosanzone1Aún no hay calificaciones
- Calculos de Soluciones 3Documento18 páginasCalculos de Soluciones 3Luis De JesúsAún no hay calificaciones
- Unidad 3. SolucionesDocumento56 páginasUnidad 3. Solucionesbillybastidas2Aún no hay calificaciones
- Disoluciones (Parte I)Documento62 páginasDisoluciones (Parte I)samgonzalez931Aún no hay calificaciones
- SolucionesDocumento50 páginasSolucionesSam menAún no hay calificaciones
- Unidad Temática 6Documento8 páginasUnidad Temática 6Cande AntónAún no hay calificaciones
- 10 Disoluciones VfinalDocumento75 páginas10 Disoluciones Vfinallucioricci012Aún no hay calificaciones
- Práctica 1 Química AnalíticaDocumento8 páginasPráctica 1 Química AnalíticaGenesis Fajardo IgotsevenAún no hay calificaciones
- Soluciones QuimicasDocumento8 páginasSoluciones QuimicasRommer HernandezAún no hay calificaciones
- Módulo 04 Química IIDocumento41 páginasMódulo 04 Química IIRocuo pyropeAún no hay calificaciones
- Química General: SolucionesDocumento27 páginasQuímica General: SolucionesGIAN GABRIEL MAZA MIOAún no hay calificaciones
- Tema 3Documento64 páginasTema 3Francisco GonzalezAún no hay calificaciones
- 8) SolucionesDocumento18 páginas8) SolucionesDiego Garcia50% (2)
- Soluciones IdealesDocumento15 páginasSoluciones IdealesYeituko MendozaAún no hay calificaciones
- 21soluciones o Disoluciones QuímicasSDocumento45 páginas21soluciones o Disoluciones QuímicasSDaniel RamosAún no hay calificaciones
- Semana 10. SolucionesDocumento39 páginasSemana 10. SolucionesLeguia GuzmanAún no hay calificaciones
- Solvente.: Soluto Solvente Mezcla Soluto + Solvente Mezcla SoluciónDocumento2 páginasSolvente.: Soluto Solvente Mezcla Soluto + Solvente Mezcla Solución766zrd6py4Aún no hay calificaciones
- Soluciones 2017-IIDocumento25 páginasSoluciones 2017-IIbrayerAún no hay calificaciones
- Quimica Soluciones - UnmsmDocumento39 páginasQuimica Soluciones - UnmsmPRINS JEREMY VILLEGAS JULCAAún no hay calificaciones
- 6a SolucionesDocumento22 páginas6a SolucionesAgustín De ZanAún no hay calificaciones
- Quimica General U10 VirtualDocumento32 páginasQuimica General U10 VirtualalejandraamartinezqAún no hay calificaciones
- Unidad No.5 Teoria de Quimica Soluciones y Solubilidad 2020-1Documento42 páginasUnidad No.5 Teoria de Quimica Soluciones y Solubilidad 2020-1Elixandra Rodriguez De FernándezAún no hay calificaciones
- Propiedades ColigativasDocumento5 páginasPropiedades ColigativaspanteraleokrugeriAún no hay calificaciones
- Estado Liquido y SolidoDocumento42 páginasEstado Liquido y Solidomilva naufamerAún no hay calificaciones
- Apuntes de SolucionesDocumento8 páginasApuntes de SolucionesRoberto SantiagoAún no hay calificaciones
- Pregunta 02 - Parcial 20211Documento1 páginaPregunta 02 - Parcial 20211JadeAún no hay calificaciones
- Curso Topicos 2022Documento11 páginasCurso Topicos 2022JadeAún no hay calificaciones
- Silabo Uni Calculo Vectorial Uni 2021-IDocumento4 páginasSilabo Uni Calculo Vectorial Uni 2021-IJadeAún no hay calificaciones
- Semana+8+ +S+Pruebas+Chi Cuadrado+ +Prueba+de+Homogeneidad1Documento22 páginasSemana+8+ +S+Pruebas+Chi Cuadrado+ +Prueba+de+Homogeneidad1JadeAún no hay calificaciones
- Lamina 6 - NeumaticaDocumento1 páginaLamina 6 - NeumaticaJadeAún no hay calificaciones
- Actividad de Aprendizaje - N°07Documento4 páginasActividad de Aprendizaje - N°07JadeAún no hay calificaciones
- Tarea 2 E 16jul2021Documento3 páginasTarea 2 E 16jul2021JadeAún no hay calificaciones
- Labfis1 - Secc E Inf4 Rojas PDFDocumento12 páginasLabfis1 - Secc E Inf4 Rojas PDFJadeAún no hay calificaciones
- Laboratorio 02 Cinematica y Caida LibreDocumento6 páginasLaboratorio 02 Cinematica y Caida LibreJadeAún no hay calificaciones
- Laboratorio 01Documento9 páginasLaboratorio 01JadeAún no hay calificaciones
- Traslado Interno 2018-2 - G1Documento1 páginaTraslado Interno 2018-2 - G1JadeAún no hay calificaciones
- Guia - 2018 2 Elizabeth 1 PDFDocumento22 páginasGuia - 2018 2 Elizabeth 1 PDFJadeAún no hay calificaciones
- Listagen Prueba6Documento19 páginasListagen Prueba6JadeAún no hay calificaciones
- Ejercicios Resueltos Diagrama de Fases - Daniel Gomariz - Ingeniería IndustrialDocumento20 páginasEjercicios Resueltos Diagrama de Fases - Daniel Gomariz - Ingeniería IndustrialDaniel Gomariz63% (8)
- Expo HDH Plus Petroleo IIDocumento22 páginasExpo HDH Plus Petroleo IISara GnrAún no hay calificaciones
- Catálogo Colección 2kR - Pinturas Isaval Perú SACDocumento15 páginasCatálogo Colección 2kR - Pinturas Isaval Perú SACLCM CONSULTORES SACAún no hay calificaciones
- Cparraes - Cinetica EnzimaticaDocumento47 páginasCparraes - Cinetica EnzimaticadanielAún no hay calificaciones
- Reporte# 1 de Lab. de Quimica Organica IDocumento2 páginasReporte# 1 de Lab. de Quimica Organica IMiguel A. Morillo CabreraAún no hay calificaciones
- Libro de Instrucciones PDF 1Documento52 páginasLibro de Instrucciones PDF 1francisco javier fernandez diezAún no hay calificaciones
- Problemas de AdsorcionDocumento2 páginasProblemas de AdsorcionLenin CanoAún no hay calificaciones
- Curvas de SolubilidadDocumento6 páginasCurvas de SolubilidadLaura MartinezAún no hay calificaciones
- Calcula Qué Volumen de Acido Clorhidrico Concentrado Del 32Documento1 páginaCalcula Qué Volumen de Acido Clorhidrico Concentrado Del 32Juanjo DiezAún no hay calificaciones
- Ejercicios PARTE 2 - Agosto 2022Documento38 páginasEjercicios PARTE 2 - Agosto 2022Nathalia ZamudioAún no hay calificaciones
- Ejercicios de MolaridadDocumento22 páginasEjercicios de MolaridadSigilfredo Patiño C.78% (9)
- Sublimacion: Insumos deDocumento16 páginasSublimacion: Insumos dePEDRO SANCHEZAún no hay calificaciones
- Nte Inen Iso 14713-1Documento7 páginasNte Inen Iso 14713-1Jonathan CuzcoAún no hay calificaciones
- Bohn - Friguscenter RefaccionesDocumento54 páginasBohn - Friguscenter RefaccionesMicky MHAún no hay calificaciones
- Presentacion MaceraciónDocumento42 páginasPresentacion MaceraciónAndrey RiverosAún no hay calificaciones
- DMC Ej 2Documento12 páginasDMC Ej 2agustin laraAún no hay calificaciones
- 11 Guia 3 Propiedades Coligativas PDFDocumento8 páginas11 Guia 3 Propiedades Coligativas PDFLiseth CristianoAún no hay calificaciones
- Colchones CaracterísticasDocumento4 páginasColchones CaracterísticasLudwig WittgensteinAún no hay calificaciones
- ResumenUASB Petar DiseñoDocumento36 páginasResumenUASB Petar DiseñoDavid Xforeverx DarkAún no hay calificaciones
- Extraccion Por SolventeDocumento26 páginasExtraccion Por SolventeSabina Reyes PinillaAún no hay calificaciones
- Bioquímica PDFDocumento3 páginasBioquímica PDFMadd VeraAún no hay calificaciones
- Aleaciones y Diagramas de Fase 2Documento36 páginasAleaciones y Diagramas de Fase 2ElmarcchAún no hay calificaciones
- Bloque 2Documento11 páginasBloque 2keniaAún no hay calificaciones
- Isoterma de Langmuir y FreundlinchDocumento6 páginasIsoterma de Langmuir y FreundlinchKarla Nohemí Contreras ValenzuelaAún no hay calificaciones
- Informe 5 Propiedades ColigativasDocumento13 páginasInforme 5 Propiedades ColigativasMiguel ArvizuAún no hay calificaciones
- MCB ThieleDocumento13 páginasMCB ThieleNick VasquezAún no hay calificaciones
- Reporte 4 BMDocumento5 páginasReporte 4 BMpaul carterAún no hay calificaciones
- Problemas - 2022 Catálisis Clases 1-2Documento3 páginasProblemas - 2022 Catálisis Clases 1-2Juan Esteban ZarzaAún no hay calificaciones
- 2°formulario Quimica - Prop - Coligativas 2NMDocumento1 página2°formulario Quimica - Prop - Coligativas 2NMVeronica Carrillo HernandezAún no hay calificaciones
- Pintura y Barnices ISO 12944-6Documento5 páginasPintura y Barnices ISO 12944-6sara50% (2)