
The evolution of duplicate gene expression in mammalian organs
- 1Department of Animal Ecology, Evolutionary Biology Centre, Uppsala University, S-75105 Uppsala, Sweden;
- 2Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, D-69120 Heidelberg, Germany
-
↵3 Joint senior authors
Abstract
Gene duplications generate genomic raw material that allows the emergence of novel functions, likely facilitating adaptive evolutionary innovations. However, global assessments of the functional and evolutionary relevance of duplicate genes in mammals were until recently limited by the lack of appropriate comparative data. Here, we report a large-scale study of the expression evolution of DNA-based functional gene duplicates in three major mammalian lineages (placental mammals, marsupials, egg-laying monotremes) and birds, on the basis of RNA sequencing (RNA-seq) data from nine species and eight organs. We observe dynamic changes in tissue expression preference of paralogs with different duplication ages, suggesting differential contribution of paralogs to specific organ functions during vertebrate evolution. Specifically, we show that paralogs that emerged in the common ancestor of bony vertebrates are enriched for genes with brain-specific expression and provide evidence for differential forces underlying the preferential emergence of young testis- and liver-specific expressed genes. Further analyses uncovered that the overall spatial expression profiles of gene families tend to be conserved, with several exceptions of pronounced tissue specificity shifts among lineage-specific gene family expansions. Finally, we trace new lineage-specific genes that may have contributed to the specific biology of mammalian organs, including the little-studied placenta. Overall, our study provides novel and taxonomically broad evidence for the differential contribution of duplicate genes to tissue-specific transcriptomes and for their importance for the phenotypic evolution of vertebrates.
The process of gene duplication is widely recognized as an important contributor to the phenotypic diversity of living organisms (Ohno 1970; Kaessmann 2010; Chen et al. 2013). It generates novel genomic material that can be molded through selective and neutral evolutionary processes. Upon duplication, one paralog may diverge in function, or both paralogs partition the ancestral function among them, in the process of neo- and subfunctionalization, respectively (Lynch and Force 2000; Kaessmann 2010). Duplicate genes may also be preserved by natural selection for gene dosage, enabling increased production of the ancestral gene product (Ohno 1970; Kaessmann 2010). Although a number of individual examples of gene duplicates with important novel functions have been described (for review, see Kaessmann 2010; Long et al. 2013), we still know relatively little about the dynamics of functional evolution mediated through gene duplications in mammals. Novel gene functions and associated phenotypes may arise through mutations that alter the sequence of the gene product and/or through regulatory mutations, which may affect gene expression (Necsulea and Kaessmann 2014). Notably, regulatory mutations are thought to underlie much of phenotypic evolution (King and Wilson 1975; Carroll 2008; Necsulea and Kaessmann 2014). Therefore, comparative gene expression studies may provide unique insights into the functional evolution of both old and new (duplicate) genes.
High-throughput RNA sequencing (RNA-seq) enables detailed cross-species transcriptome comparisons (Necsulea and Kaessmann 2014). However, while mammalian RNA-seq data have been used to study the evolution of 1:1 orthologous (single-copy) genes (Brawand et al. 2011; Warnefors and Kaessmann 2013; Necsulea and Kaessmann 2014) and some specific aspects of paralogs (Chen and Zhang 2012; Rogozin et al. 2014), the evolutionary and functional relevance of gene duplication still remains little explored globally, although two previous studies assessed patterns of expression evolution for subsets of duplicate gene pairs (Assis and Bachtrog 2015; Lan and Pritchard 2016). Here, we close this gap and, using an extensive RNA-seq data set, perform large-scale comparative analyses to assess short- and long-term dynamics of duplicate gene expression evolution across eight mammals and one bird. We focus on DNA-based duplications, which may arise through misguided recombination and replication processes in the germline, or through meiotic non-disjunction, in the case of whole-genome duplication (Hastings et al. 2009; Marques-Bonet et al. 2009). DNA-based duplicates constitute a major subset of duplicated genes in the genome, and we recently described expression evolution of the other major type, RNA-based duplicates, in a dedicated study (Carelli et al. 2016).
We started by studying general patterns of expression evolution and then focused on lineage-specific processes. We asked the following: (1) How do expression levels, expression divergence, and tissue specificity globally change with duplication age, and do these patterns differ by tissues or among studied species? (2) What is the significance and evolutionary role of particular tissues in the retention of duplicate genes? (3) What is the potential contribution of paralogs to lineage-specific phenotypic evolution?
Results
Re-annotation of duplicated gene families and expression level dynamics
To establish a high-confidence gene duplication data set, we downloaded protein-coding gene family trees from Ensembl v64 (Vilella et al. 2009; Flicek et al. 2012) and employed a rigorous multistep filtering procedure that removed poorly supported duplications, misannotations, and intronless genes (Supplemental Fig. S1; Supplemental Methods). Inference of duplication age was based on gene tree topology and validated by measuring the rate of synonymous substitutions (dS)—the best-suited approach for duplications across large temporal scales including ancient and recent events (Huerta-Cepas and Gabaldón 2011). The final data set contained 7350 duplication events in 4187 gene families (Supplemental Fig. S1; Supplemental Table S1). For comparisons, we also obtained a non-overlapping set of 3379 amniote single-copy orthologous genes (Methods; Brawand et al. 2011). Expression analyses were based on our previous RNA-seq data sets (Brawand et al. 2011; Necsulea et al. 2014), which comprise eight different organs (cortex or whole brain without cerebellum, cerebellum, heart, kidney, liver, testis, ovary, placenta) for eight representatives of the three major mammalian lineages (placental mammals: human, chimpanzee, gorilla, orangutan, rhesus macaque, and mouse; marsupials: gray short-tailed opossum; monotremes: platypus) and a bird (nondomesticated chicken) (Supplemental Table S2). We employed a careful read mapping and expression level estimation procedure that takes into account divergence levels of duplicate gene copies and proportions of uniquely mapped reads to infer reliable expression profiles of paralogs (Supplemental Methods). Our procedure effectively removes 91.4% of problematic human genes for which expression levels cannot be reliably determined (Supplemental Methods; Robert and Watson 2015). The few remaining genes have only been used in global analyses and therefore are unlikely to bias our results.
Global patterns of expression divergence
The neo- and subfunctionalization models of gene duplication postulate that paralogs diverge in function or partition the ancestral functions among the copies (Kaessmann 2010; Long et al. 2013), resulting in greater expression divergence and potentially higher tissue specificity in gene duplicates compared to single-copy genes. To gauge how new expression patterns emerge following gene duplication, we assessed expression divergence of paralogs of different duplication ages.
We observed an arch-shaped relationship between expression divergence (measured as Euclidean distance) (Supplemental Methods) and duplication age in all studied species (Fig. 1A; Supplemental Fig. S2). Because measures of expression divergence can be affected by the presence of tissue-specific expressed genes (Chen and Zhang 2012), we confirmed that the observed pattern was robust to the removal of tissue-specific expressed, nonexpressed, or lowly expressed genes, with the sum of expression across all tissues <1 FPKM (fragments per kilobase of exon per million of mapped reads) (Supplemental Fig. S3). A similar, albeit considerably less pronounced, pattern was observed with an alternative measure of expression divergence, Pearson's correlation coefficient (r) (Fig. 1B; Supplemental Fig. S4). As expected, the youngest paralogs showed low expression divergence, likely because their short time of independent evolution was not sufficient for them to diverge in expression. However, while genes from the younger age classes showed progressively higher expression divergence with age, as previously observed for a subset of these data (Assis and Bachtrog 2015), the pattern was reversed for paralogs that predate the emergence of tetrapods. In these older genes, expression divergence decreased with age, so that the oldest and the youngest paralogs showed a similar degree of expression divergence. High expression levels of old duplicates may partly explain their low expression divergence: Expression divergence is negatively correlated with expression levels in paralogs and single-copy genes (rho = −0.35 and −0.47, P < 10−15, for human-mouse single-copy genes and all studied paralogs, respectively) (Supplemental Fig. S5) and old paralogs are highly expressed (Fig. 2). Generally, expression levels and duplication age are correlated in paralogs (rho = 0.18–0.35 in all studied species, P < 10−15) (Fig. 2). This relationship persists after removing lowly expressed genes with the sum of expression <1 FPKM across all tissues (rho = 0.16–0.31, P < 10−15) and even within individual gene families (rho = 0.22, P < 10−15).

Expression divergence of single-copy human-mouse orthologs (yellow) and age-grouped mouse paralogs (blue) based (A) on Euclidean distances and (B) on Pearson's correlation coefficient r (displayed as 1−r). Paralogs are grouped into age classes according to gene tree topology. (C) The species tree shows divergence times in million years, with highlighted branches corresponding to the evolutionary groups for which divergence was inferred.

Median expression levels for single-copy (yellow) and duplicate genes (shades of blue) across all organs in the mouse genome. Duplicate genes are grouped into age classes.
Estimates of expression divergence with Euclidean distances take into account expression levels (Pereira et al. 2009). Recent studies in Paramecium and mammals highlighted the importance of considering expression levels by demonstrating rapid emergence of significantly asymmetric expression levels between paralogs (Gout and Lynch 2015; Lan and Pritchard 2016). In contrast, Pearson's r estimates reflect spatial patterns of expression divergence without accounting for differences in expression levels. Pearson's r also tends to overestimate expression divergence for uniformly expressed genes, whereas Euclidean distances are robust to such expression patterns (Pereira et al. 2009). It is therefore possible that expression divergence for ancient, more ubiquitously expressed paralogs has been overestimated with Pearson's r in our data set, masking the arch-shaped relationship between expression divergence and duplication age (Fig. 1B).
Expression divergence of paralogs of any duplication age was higher than that of human-mouse single-copy genes (Fig. 1A), even if the paralogs were evolutionarily younger (P < 10−14) (Supplemental Fig. S6), confirming that paralogs diverge in expression soon after duplication (Chen and Zhang 2012; Rogozin et al. 2014; Assis and Bachtrog 2015). However, paralogs are generally expressed at significantly lower levels than single-copy genes in all studied species (P < 10−13, Kruskal–Wallis test) (Fig. 2), and expression levels of young paralogs are particularly low. To evaluate if higher expression divergence of paralogs is the result of technical and/or biological noise typical for lowly expressed genes, we performed a global test for the contribution of expression level, evolutionary time (duplication or speciation age), and duplication status (duplicate or single-copy genes) on expression divergence. We used mouse and human data, for which the presence of other rodent and primate genomes allowed more fine-grained duplication and speciation age estimates. Because expression divergence was independent of the number of gene copies (including copies stemming from subsequent rounds of duplication/speciation) that could be traced back to a given duplication event (Spearman's rho = 0.019, P = 0.33 and rho = −0.005, P = 0.81 for mouse and human, respectively), we did not include this factor in the model. We found that expression levels explained most of the variance in expression divergence, followed by evolutionary time and duplication status (Methods; Supplemental Table S3). Our results thus show that global differences in expression levels explain much of the difference in expression divergence between duplicated and single-copy genes. Using Pearson's r, the global model of expression divergence evolution had less explanatory power and the effect of expression levels was smaller than the effect of evolutionary time, consistent with the insensitivity of Pearson's r to differences in expression levels (see above; Supplemental Table S4).
Expression divergence and protein sequence divergence are correlated in paralogs
Two types of mutations underlie the evolution of novel gene functions: changes to the protein sequences and changes to gene expression (Necsulea and Kaessmann 2014). Expression divergence is believed to be acquired more rapidly than sequence divergence (Wapinski et al. 2007), but both were found to be correlated in 1:1 orthologs (Khaitovich et al. 2005; Warnefors and Kaessmann 2013) and in paralogs that have diverged little on sequence level (Gu et al. 2002; Makova and Li 2003; Nehrt et al. 2011; Liao et al. 2014), suggesting considerable coupling of these processes. Consistently, we observe weak but significant global correlation between expression divergence (Euclidean distances) and protein sequence divergence (nonsynonymous substitution rates, dN) in paralogs of two species (human, as the representative of placental mammals, and opossum, as the representative of marsupials) (Fig. 3A). The correlation was strongest for young paralogs and systematically decreased with duplication age. Expression divergence levels off during amniote evolution and decreases for evolutionarily older genes (Fig. 1A), whereas protein sequence divergence continues to increase (Brawand et al. 2011; Warnefors and Kaessmann 2013). Therefore, reduced correlation between expression and protein sequence divergence for ancient and old paralogs can be expected, although it is still detectable even across large evolutionary scales.

Expression divergence and protein sequence divergence. (A) Correlation between expression divergence and protein sequence divergence for human and opossum paralogs of different duplication ages and for 1:1 amniote and primate orthologs (Warnefors and Kaessmann 2013). Numbers of gene families within each group are shown above the bars. A single gene pair was sampled for each gene family and expression divergence was measured as Euclidean distances. (*) P < 0.02, (**) P < 0.001, (***) P < 0.00001, (NS) not significant. (B) Expression divergence (measured as Euclidean distances across all organs) and dN values for human duplicate genes of all duplication ages. Increasing bias toward expression divergence is indicated in shades of yellow and increasing bias toward protein divergence in blue. (C) Relationships among the five most overrepresented GO terms for ED-biased (orange) and dN-biased (blue) genes in each age category. Brighter colors correspond to young and lineage-specific duplication ages, darker colors to old and ancient duplication ages. Some intermediate terms were omitted for clarity.
Some genes are likely to diverge more in terms of expression than in terms of sequence, and vice versa. Among 1:1 orthologs, genes escaping the global correlation were found to fulfill different functions (Warnefors and Kaessmann 2013). To investigate if similar functional variation can be found in paralogs, we performed Gene Ontology (GO) enrichment analyses, dividing human paralogs into those that diverged more on the protein sequence level relative to their expression divergence (termed “dN-biased”) and those that diverged more in expression profiles relative to protein sequence (“ED-biased”) (Fig. 3B; Methods; Warnefors and Kaessmann 2013). Because the correlation between expression and sequence divergence changes with evolutionary time (Fig. 3A), we grouped the paralogs into two duplication age categories: young paralogs that duplicated in the common amniote ancestor or more recently and old paralogs that duplicated in the common tetrapod ancestor or earlier. Similar to findings in 1:1 orthologs, we observed clear functional differences between dN- and ED-biased paralogs, with a considerable effect of duplication age. Young dN-biased paralogs shared with dN-biased 1:1 orthologs enrichment for genes implicated in transcription and regulation of gene expression (Supplemental Table S5; Fig. 3C; Warnefors and Kaessmann 2013). They shared involvement in metabolic and biosynthetic processes with old dN-biased genes (Supplemental Table S5; Fig. 3C). We also detected considerable functional agreement between old ED-biased paralogs and ED-biased 1:1 orthologs: They were involved in synaptic transmission, cell signaling and communication, ion transport, and anatomical structure development (Supplemental Table S5; Fig. 3C). However, young ED-biased paralogs were implicated in functions not observed among biased 1:1 amniote orthologs, such as metabolic and cellular processes, with “xenobiotic metabolism” being the most significant term (adjusted P = 6 × 10−10 after correction for multiple testing; enrichment score = 1.92) (Supplemental Table S5; Fig. 3C). This finding is consistent with the high proportion of liver-specific expressed genes among young paralogs (see below), strengthening the role of gene duplicates as sources of evolutionary novelty and emphasizing their contribution to phenotypic evolution. Liver-expressed genes involved in detoxification and waste removal were found to show lineage-specific expression changes among amniote orthologs (Brawand et al. 2011) and pronounced inter-individual variation in expression levels (Khaitovich et al. 2005), possibly reflecting regulatory plasticity, which may allow for more rapid expression evolution (Romero et al. 2012).
Tissue-specific functional contributions of paralogs of different ages
The expression patterns of paralogs have generally been found to be more tissue-specific than those of single-copy genes (Huminiecki and Wolfe 2004; Huerta-Cepas and Gabaldón 2011), in line with the neo- and subfunctionalization models of gene duplication. However, to understand why tissue specificity is elevated in paralogs, it is necessary to study the contribution of expression levels, evolutionary time, and duplication status in a common framework. We thus ran linear models using two alternative measures of tissue specificity: (1) tau (Yanai et al. 2005); and (2) calculating tissue specificity as the relative expression of the gene in the tissue with highest expression (Methods). Expression levels explained most of the variance in tissue specificity, whereas evolutionary time was not significant (Supplemental Tables S6, S7). Paralogs showed more pronounced tissue-specific expression than single-copy genes in the model based on tau (Supplemental Table S7), in concordance with previous studies (e.g., Huminiecki and Wolfe 2004; Huerta-Cepas and Gabaldón 2011). As we detail below, high tissue specificity of lowly expressed genes bears biological relevance and cannot be explained by technical/biological noise alone. However, the overall low fit of these models suggests that patterns of tissue expression specificity might be best explained by other factors (e.g., tissue complexity). Nevertheless, combined with our analyses of expression divergence, this result supports the notion that the expression level of a gene represents a strong predictor for explaining the dynamics of evolutionary expression pattern divergence.
We speculated that the contributions of duplicated genes to the emergence of novel functions might be reflected in the tissue-specific expression of paralogs that originated at different evolutionary times. Indeed, we observed consistent, substantial, and statistically significant differences in the distribution of tissue preferences by duplication age in all studied species (Fig. 4; Supplemental Fig. S7). They were not affected by our definition of tissue specificity (tau, “stringent,” or “twice uniform expression”) (Methods), expression levels, separate or combined treatment of neural tissues, or whether the analyses were carried out at the gene or gene family level (Supplemental Fig. S8).

Evolutionary dynamic expression profiles of tissue-specific expressed genes. (A) Schematic representation of duplication age categories using opossum as an example. (B) Proportion of genes specific to any given tissue is plotted by gene type and duplication age. Tissue specificity of opossum genes was assessed using the “stringent” definition (see Methods). Single-copy genes and paralogs, grouped in four age classes, are shown for each tissue. Bars represent 95% confidence intervals. Analyses carried out at gene level. (C) Significant differences in evolutionary dynamics of tissue preferences of duplicated genes. Duplication ages are given in the upper row. Bars above group the fine-grained duplication ages into the same four age categories as in A (Methods). Significant over- (blue) or underrepresentation (orange) of genes with highest expression in a given tissue was tested for paralogs in each species and for each duplication age with a χ2 test, followed by a post hoc procedure (Methods). Gray cells signify no statistical difference. (D) Analysis of differential gene loss/gain in gene families with highest expression in liver, testis, and all other tissues combined. (E) Expression shift analysis: Proportions of chicken outgroup genes with highest expression in liver and testis compared to the proportion of resulting mammalian paralogs with highest expression in these tissues. Significance was assessed with a χ2 test.
The proportions of lineage-specific and young heart- and particularly brain-specific expressed paralogs were significantly lower than expected (Fig. 4B,C; Supplemental Figs. S7, S8), suggesting that young duplicates have contributed proportionally less to the functional evolution of heart and brain tissues. Across all expression levels, young paralogs showed low relative expression in heart and neural tissues compared to older paralogs (Supplemental Methods; Supplemental Figs. S9, S10). Notably, the highest proportion of brain-specific expressed genes was among old paralogs that duplicated in the common bony vertebrate ancestor (Fig. 4B; Supplemental Figs. S7, S8)—an evolutionary time period marked by substantial elaborations in the morphology and cyto-architectural complexity of the telencephalon and an overall increase in brain-to-body size ratio (Butler 2010). Thus, our large-scale survey suggests that gene duplications may have facilitated these phenotypic vertebrate brain innovations, consistent with a previous analysis of individual gene families (Chen et al. 2011).
The proportion of testis-specific expressed paralogs was significantly higher than expected among lineage-specific and young duplicates (Fig. 4B,C; Supplemental Figs. S7, S8). Testis-specific expressed single-copy and duplicate genes of all ages tend to be overall lowly expressed (Supplemental Fig. S11), consistent with the idea of widespread spurious transcription in this tissue (Soumillon et al. 2013). However, despite low expression, lineage-specific and young testis-expressed paralogs are significantly enriched for genes functional in gamete generation, reproductive processes, and spermatogenesis (retinoic acid pathway) compared to the background of all testis-expressed genes (Benjamini–Hochberg-corrected P < 10−3) (Supplemental Table S8). Thus, they are not merely a product of the permissive transcriptional environment (Soumillon et al. 2013) but may have contributed important testis-specific functions during more recent mammalian evolution.
Lineage-specific and young duplications also contained higher than expected proportions of genes specifically expressed in the liver (Fig. 4B,C; Supplemental Figs. S7, S8). However, in contrast to the testis, liver-specific genes tend to be highly expressed, so that relative liver expression increased with expression levels in single-copy and duplicate genes (Supplemental Fig. S12). Young liver-specific expressed paralogs showed functional enrichments for metabolic and catabolic processes related to digestion and detoxification (Benjamini–Hochberg-corrected P < 10−3) (Supplemental Table S8), signifying their contribution to the typical liver-associated functions over and above other liver-expressed genes.
Evolutionary forces underlying the preferential emergence of testis- and liver-expressed genes
The high proportion of young testis- and liver-specific expressed genes could be the result of two not mutually exclusive processes: rapid gene turnover (i.e., increased duplication fixation rate in young paralogs, followed by gene loss later in evolution) and/or changes in expression profiles with evolutionary time, so that young genes become more broadly expressed with time or shift their expression to another tissue. To study the contribution of these processes, we first quantified the rate of gene gain and gene loss in human and mouse paralogs following a duplication in the common human-mouse ancestor (within the young age class) (Methods). We chose these two species as they have the highest-quality genomes among our study species, which reduces biases from wrongly inferred gene losses or spurious duplicates. Gene families that have experienced gene losses were identified as having fewer than four paralogs in both species together, whereas gene families that have experienced gene gains had more than four mouse and human paralogs in total. We established the major tissue of expression for each gene family by determining the tissue with highest median expression across all paralogs. A global test showed significant differences in the numbers of gene families that were dominated by gene losses and gene gains among gene families with predominant expression in liver, testis, and all other tissues combined (χ2 = 9.77, df = 2, P = 0.008). A post hoc procedure (Methods) revealed that gene families with highest expression in the liver had significantly more gains and fewer losses following a duplication in the common human-mouse ancestor (Fig. 4D). This finding suggests that new gene copies from liver-expressed gene families tend to be fixed more often than gene copies from families expressed in other tissues, thus contributing to the high proportion of young and lineage-specific liver-expressed paralogs. The proportions of gains and losses did not differ between testis-expressed gene families and gene families expressed in other tissues (Fig. 4D), suggesting that a different process leads to increased proportions of young testis-specific expressed genes.
Thus, we next tested for changes in expression profiles in testis- and liver-expressed young paralogs, focusing on a subset of duplication events in mammals, for which a single-copy chicken gene could be determined as an outgroup (n = 821, containing 2662 paralogs in all mammalian species included in our study). We hypothesized that if expression profiles are preserved following recent duplication, the proportion of single-copy outgroup chicken genes with highest expression in liver and testis should be the same as the proportion of mammalian paralogs with highest expression in these tissues. Indeed, we observed no difference in the proportion of single-copy outgroup chicken genes and mammalian paralogs with highest expression in liver (χ2 = 0.08, df = 1, P-value = 0.78) (Fig. 4E). However, we found a greater proportion of mammalian paralogs with highest expression in testis compared to chicken outgroup genes (χ2 = 6.45, df = 1, P-value = 0.01) (Fig. 4E), indicating that young paralogs tend to be testis-expressed but change their expression pattern with evolutionary time.
Taken together, our results suggest that different processes are responsible for the high proportion of young and lineage-specific testis- and liver-specific expressed paralogs. Fast gene turnover in liver-expressed gene families could provide an important mechanism for rapid, lineage-specific dietary adaptations, together with species-specific changes in expression of liver-specific genes, which were suggested to be linked to ecological adaptations in primates (Perry et al. 2012). In contrast, the evolution of testis-expressed gene families predominantly involves shifts and/or broadening of expression profiles with evolutionary time. This observation is in agreement with the “out of the testis” hypothesis of gene origination (Kaessmann 2010), making it a shared mechanism for DNA- and RNA-based gene duplications (Carelli et al. 2016; see also Assis and Bachtrog 2015). Young testis-expressed paralogs may directly contribute to lineage-specific biology as evidenced by their involvement in reproductive functions despite low expression levels (see above; Supplemental Table S8). Reproductive proteins show rapid sequence evolution in animals (Swanson and Vacquier 2002), and our findings support the notion that gene duplications contribute to species-specific reproductive characteristics (Clark et al. 2007; Almeida and Desalle 2008; Kelleher and Markow 2009; Kaessmann 2010; Betrán 2015). Overall, our results illustrate how gene duplications from different evolutionary time periods have contributed differentially to the transcriptomes and functions of various mammalian/amniote organs.
Gene duplications and the emergence of the placenta
The placenta is an evolutionarily young tissue and the most varied mammalian organ, with physiological and anatomical homologies among marsupials and eutherians (Renfree 2010; Wildman 2016). We hypothesized that new duplicate genes might have been recruited into the placenta during the establishment of this organ, thus providing raw material for the evolution of placenta-specific functions. However, instead we found that older paralogs with duplication ages in mammals and amniotes contained significantly more genes with placenta-specific expression than expected, whereas paralogs that duplicated in the common therian and eutherian ancestors did not show increased numbers of placenta-specific expressed genes (Fig. 5). Thus, paralogs that predate the placenta emergence appear to have been co-opted for functions in this tissue, in line with previous work (Knox and Baker 2008). Placental morphology and physiology are highly varied in therian mammals (Renfree 2010; Wildman 2016), and this organ has repeatedly recruited genes with similar functions but evolutionarily independent origins, e.g., the syncytin-like genes in primates, rodents, and lagomorphs (Mi et al. 2000; Dupressoir et al. 2009; Heidmann et al. 2009). It is thus conceivable that new and old genes acquired functional roles ever since the emergence of the placenta in a lineage-specific manner.

Placenta-enrichment by duplication age in three therian species: human, mouse, and opossum. Significant global χ2 tests indicate differences in the number of placenta-specific expressed genes by age group in each of the studied species (placenta-specific expressed genes are defined as having more than twice uniform expression level in the placenta), after which a post hoc procedure was applied, as described in Methods. (+) Significant overrepresentation of placenta-specific expressed genes, (–) significant underrepresentation of placenta-specific expressed genes, (NS) not different from expectation. Overall, results obtained with the alternative definition of tissue specificity (tau) were the same, with the exception of mouse and human, where lineage-specific paralogs showed more placenta-specific expressed genes than expected.
We identify well-known genes that have acquired placenta-specific expression following gene duplication (e.g., IGF2 [Supplemental Table S1, genefam_3056], and HBG1 and HBG2 [Supplemental Table S1, genefam_1850]). However, the most extreme example of placenta-specific expression gain following duplication is PAGE4 (P antigen family, member 4), a member of the GAGE/PAGE gene family (Fig. 6; Supplemental Table S1, genefam_2135). Homologs of PAGE4 show moderate expression levels (1–95 FPKM) specifically in the testis and have experienced multiple rounds of duplication. In contrast, PAGE4 acquired high levels (>2600 FPKM) of placenta-specific expression in the human lineage, following a duplication event in the common eutherian ancestor (Fig. 6). The entire gene family appears to be missing in the mouse, but PAGE4 is expressed in the elephant placenta (Hou et al. 2012). Previous studies showed that PAGE4 is also expressed in other reproductive organs and cancer cell lines (Iavarone et al. 2002) and thus seems to be associated with fast-proliferating cells. It has received attention as a cancer/testis antigen that is up-regulated in prostate cancer and is involved in the stress-response pathway linked to prostatic development and disease (Mooney et al. 2014). However, its expression patterns suggest that it may fulfill an important placenta-specific function in primates and possibly other eutherians. In mice, its role may be taken by a different gene or set of genes. Altogether, we can pinpoint interesting candidate paralogs with extreme shifts in expression profiles which potentially signal their functional relevance in a newly emerged tissue.

PAGE4 gene family tree. Duplication events are marked with blue squares. Gene names, Ensembl gene ID, and the species are given at the tips of the tree. Circles represent expression patterns, with circle color corresponding to the tissue of highest expression of the respective gene and circle size approximating the expression level. It is likely that homologs of PAGE4 are placenta-specific expressed in other eutherian species, as suggested by the study in the elephant (Hou et al. 2012); however, we could not assess this pattern, as placenta samples from other primates were not available to our study. The absence of expression of PAGE3 and PAGE5 in orangutan is likely explained by the unavailability of testis samples from this species to our study.
Spatial expression dynamics and lineage-specific expansions of amniote gene families
To better understand the contribution of duplicated genes to phenotypic evolution, we set out to assess how tissue-specific expression profiles of amniote gene families evolve following repeated duplications. We characterized expanded amniote gene families into those containing only broadly expressed paralogs and those containing primarily tissue-specific expressed paralogs. Among tissue-specific expressed gene families, we investigated how often specificity for the same tissue is preserved in repeated, independent duplications along individual amniote lineages.
We used twice uniform measure of tissue specificity to classify gene families in each of our study species into: (1) “broad” gene families, in which all members are expressed at comparable levels in all tissues (0%–8.9% of gene families by species); (2) “diverse” gene families, which contain paralogs specific for different tissues (26.0%–51.2%); and (3) “specific” gene families, in which the majority of paralogs are specific for the same tissue (37.7%–58.6%) (Table 1; Methods). Using tau as the alternative measure of tissue specificity, the results were similar, although fewer gene families were classified as “specific” and “diverse” and more as “broad” (Supplemental Table S9).
Expression dynamics of anmiote gene families
We found that “broad” gene families frequently contain zinc-finger genes (43%–100%) (Table 1; Supplemental Table S9), in accordance with the universal role of these genes in regulating fundamental processes, such as transcription. Species-specific expansions of KRAB zinc-finger gene families are well characterized in eutherians (Emerson and Thomas 2009) and marsupials (Goodstadt et al. 2007). These genes are implicated in expression control of mobile genetic elements during development (Rowe and Trono 2011) and in adult tissues (Ecco et al. 2016). However, assessing the individual expression patterns of many young zinc-finger paralogs is hampered by the difficulty of distinguishing transcribed copies from a pool of closely related paralogs.
Next, we asked how often gene families preserved their expression profile after experiencing multiple independent duplications along individual lineages (Fig. 7; Supplemental Table S10; Methods). Considering human, mouse, opossum, platypus, and chicken, we identified a total of 25 gene families that expanded independently in two or more species (Fig. 7). Only two gene families expanded independently in more than two species. One of them duplicated in mouse, opossum, and platypus and contained butyrophilin and its paralogs that are involved in adaptive immune response and lipid, fatty acid, and sterol metabolism, including variants linked to metabolic syndrome. It showed specific expression in mouse testis and opossum liver but was “diverse” in platypus. The other gene family duplicated independently in mouse, opossum, platypus, and chicken and contained guanylate binding peptides that are involved in immune response against different classes of pathogens. Opossum and chicken paralogs were specifically expressed in ovary, whereas mouse and platypus paralogs showed a wide range of tissue specificities (Fig. 8A).

Lineage-specific and shared gene family expansion in amniotes. (A) Gene family expansions along primate, rodent, marsupial, monotreme, and bird lineages are depicted along the tree branches as bar-plots (number of gene families with at least three paralogs, number of “specific,” number of “diverse,” and number of “broad” gene families). (B) Gene families that expanded independently in more than a single lineage, with gene family identifiers depicted above (as in Supplemental Table S1). Color of the cells corresponds to the predominant tissue of expression of each gene family in the given species for “specific” gene families. White cells correspond to “diverse” gene families. The gene family highlighted in red above expanded independently in mouse, opossum, platypus, and chicken; the blue gene family expanded independently in mouse, opossum, and platypus.
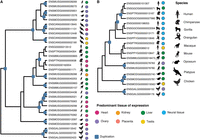
Lineage-specific expansion and expression changes in amniotes. Duplication events are marked with blue squares. Ensembl gene IDs and species are shown at the tips of the trees. Circles represent expression patterns, with circle color corresponding to the tissue of the highest expression of the respective gene. (A) The gene family (Fam612) containing guanylate binding peptides has expanded independently in mouse, opossum, platypus, and chicken. Note multiple lineage-specific changes in tissue preference. (B) Independent lineage-specific duplications in primates and marsupials. Expansions along the primate lineage produced paralogs with high brain-specific expression, whereas the majority of opossum genes show low liver-specific expression.
Among the 23 gene families that expanded independently in only two species, none were “broad,” seven were “diverse” in both species, eight were “diverse” in one but “specific” in the other species, and eight were “specific” in both species (Fig. 7B). There was a tendency to preserve specificity for the same tissue (six of the eight events) (Fig. 7B). In one of the “specific” gene families that differed in tissue specificity between the two species, expansions along the primate lineage produced brain-specific expressed paralogs, whereas paralogs on the marsupial lineage showed preference for liver expression (Fig. 8B). Human paralogs of the aforementioned gene family are known to be functional in brain development and possibly synaptogenesis (e.g., SIRPA) (UniProt Consortium 2015), supporting the idea that young duplicates have an important role in species-specific phenotypes. Our analyses thus indicate that lineage-specific expansions of the same gene family can evolve unique expression patterns in different lineages, highlighting the dynamic nature and functional flexibility of gene duplicates.
Discussion
Gene duplications are unequivocally recognized as an important source of evolutionary novelty (Kaessmann 2010; Chen et al. 2013). Here, using comparative RNA-seq data from a comprehensive set of nine amniotes, we conducted an in-depth study of evolutionary dynamics of gene expression changes following DNA-based duplications.
Divergent expression profiles and increased tissue specificity are frequently considered to be the hallmarks of gene duplication (Conant and Wolfe 2008; Huerta-Cepas et al. 2011). In this study, we confirm that paralogs are generally more divergent in expression profiles and more tissue-specific than single-copy genes and that expression divergence is acquired quickly after gene duplication. We also highlight the overall pronounced difference in expression levels between single-copy and duplicated genes and that the expression level of a gene represents a strong predictor of evolutionary expression pattern divergence. A combination of various factors may explain these observations. Gene duplications can directly lead to reduced expression levels of resulting copies, either through incomplete duplication of regulatory elements or due to a special form of subfunctionalization, in which expression reduction facilitates paralog retention (Qian et al. 2010). Indeed, rapid reduction in expression levels was demonstrated in human paralogs that emerged since the human-macaque split (Lan and Pritchard 2016). Also, lowly expressed nonessential gene families may duplicate and be retained after duplication more readily, and young paralogs were shown to be enriched for nonessential genes (Woods et al. 2013; Grishkevich and Yanai 2014), providing a link between gene age and expression levels. The overall rapid expression divergence of duplicate gene copies is likely explained by the frequent change in the regulatory landscape following gene duplication (i.e., the loss and gain of regulatory elements) (see also above), the reduced selective constraint afforded by the availability of an extra gene copy (Ohno 1970), and the reduced selective constraint due to the generally lower expression levels of duplicates (COSTEX model) (Gout et al. 2010). The latter likely also explains the rapid divergence of lowly expressed single-copy genes. In summary, gene expression levels are an important factor for understanding the dynamics of functional divergence and emergence of evolutionary novelty.
Despite generally low expression levels of young paralogs, our analyses indicate that they can be functionally relevant (Supplemental Table S8). We further show that expression levels systematically increase with evolutionary time, so that old paralogs are expressed at higher levels than young paralogs (Fig. 2). It is worthwhile to note the large variance in expression levels of young and lineage-specific paralogs, in agreement with increased expression asymmetry of genes with these evolutionary ages (Gout and Lynch 2015; Lan and Pritchard 2016). However, contrary to our findings, no differences in expression levels were found in yeast paralogs of different evolutionary ages (Qian et al. 2010). The yeast study considered older evolutionary branches than the duplication ages analyzed here, and it is possible that old paralogs reach a plateau, beyond which the increase in expression levels is only marginal. Indeed, we observe little change in expression level in duplications older than the common vertebrate ancestor (535 Mya) (Fig. 2). Furthermore, inherent biological factors (e.g., unicellularity) may prevent changes in expression levels of yeast paralogs, or these changes may have remained undetected due to low sample size of analyzed yeast paralogs of any given evolutionary age (Qian et al. 2010).
The pattern of increasing expression divergence with duplication age for young paralogs (Fig. 1) is highly consistent with the recent study by Lan and Pritchard (2016), who demonstrated that chromosomal separation of paralogs is crucial for acquisition of independent expression profiles and that this separation is achieved gradually with evolutionary time. Hence, as chromosomal rearrangements decouple gene expression regulation of initially tandem duplicates, their expression profiles diverge. In contrast, for old, highly expressed paralogs, reduced expression divergence is expected under the COSTEX model (Gout et al. 2010). In addition, all ancient genes have experienced two rounds of whole-genome duplications in the common vertebrate ancestor (Dehal and Boore 2005). This process, by duplicating the entire genomic content, preserves stoichiometric relationships between gene products, and the resulting paralogs experience increased selection against changes in expression and copy number, in the case of dosage-sensitive genes (Makino and McLysaght 2010; McLysaght et al. 2014). These two processes, one specific to young paralogs and the other dominant in old paralogs, reconcile the observed arch-shaped distribution of expression divergence observed in our data (Fig. 1).
Our set of study tissues includes representatives of all three germ layers and covers major internal organs. However, different tissues are affected by different biological and evolutionary processes, as exemplified by our finding of dynamic changes in the proportions of tissue-specific expressed genes through time. Therefore, future work that includes a larger tissue collection is needed for a thorough exploration of the anatomic complexity of amniotes. For instance, additional organs involved in digestion (e.g., stomach, and pancreas) could help refine the suggested role of new genes in dietary adaptations. Our tissue selection was inadequate to study single coding exon olfactory genes that are enriched for young duplicates and contained within heavily expanded gene families (Young and Trask 2002; Nei et al. 2008). This task will require dedicated RNA expression profiling of olfactory (sensory) tissues.
Our analyses consistently point to the importance of taking into account tissue complexity when studying the contributions of genes to organismal diversity. For instance, we found many old paralogs that show low levels of brain-specific expression but are enriched for functions related to synaptic transmission, cognition, learning, and memory (Supplemental Table S11), which suggests that their preferential expression in the brain is genuine and biologically relevant. Because we analyzed bulk tissues, genes that are specifically expressed in certain regions or cell types will show high tissue specificity but low expression. In the future, it will thus be important to systematically study transcriptional differences between cell types and tissue regions (Hawrylycz et al. 2012).
Another important aspect to be considered in future studies is the changing dynamic of duplicate gene expression during development. For instance, our analyses of adult brain suggest that young paralogs contribute little to the brain-specific transcriptome, whereas Zhang et al. (2011) found young paralogs to be specifically expressed in the developing human brain. A study that tracked changes in gene expression during placentation showed how paralogs of different duplication ages are expressed at different stages of pregnancy (Knox and Baker 2008). It is likely that such dynamics will be observed in many, if not most, organs.
Taken together, our study provides a comprehensive evolutionary analysis of amniote gene families in a comparative manner, spanning a large evolutionary time scale. We describe general features of duplicate evolution, which allow pinpointing cases with unique trajectories and therefore potentially lineage-specific adaptations. The stringently filtered database of gene duplications and associated expression values allows exploring lineage-specific shifts in expression profiles that might be indicative of evolutionary innovations and identifying interesting candidate genes with specific characteristics that merit experimental evaluation.
Methods
Duplication and single-copy genes data sets
From the initial set of protein-coding gene family trees that was obtained from Ensembl v64 (Vilella et al. 2009; Flicek et al. 2012) we retained only those gene family trees (n = 12,452, number of duplication events = 18,859) that contained at least one of the species for which transcriptome data were available (Supplemental Fig. S1). To retrieve and annotate duplication events, we relied on ETE v2 (Huerta-Cepas et al. 2010). Each gene family tree was parsed from the root to the leaves. Upon encountering a duplication event, the daughter clades were analyzed and a number of filtering steps employed to remove poorly supported duplication nodes, duplications with incorrectly inferred duplication ages, erroneously inferred duplications (stemming from split genes or based on transcriptional evidence with overlapping coordinates), and intronless gene copies (Supplemental Methods).
A data set of 1:1 amniote orthologs was taken from Brawand et al. (2011). However, because we also considered duplications with ages older than the common amniote ancestor, we removed genes that were present in our duplication data set from the single-copy data set. In total, we retained 3379 single-copy genes.
Expression data
RNA-seq expression data were available for nine species, belonging to the three main mammalian lineages (placental mammals: human, chimpanzee, gorilla, orangutan, rhesus macaque, mouse; marsupials: gray short-tailed opossum; monotremes: platypus) and a bird (nondomesticated chicken) from five somatic (cortex or whole brain without cerebellum, cerebellum, hear, kidney, liver) and three reproductive tissues (testis, ovary, placenta) (Gene Expression Omnibus accession numbers GSE30352 [Brawand et al. 2011] and GSE43520 [Necsulea et al. 2014]) (Supplemental Table S2). Adapter-trimmed RNA-seq reads were aligned on the reference genomes with TopHat (Trapnell et al. 2009), and gene expression was estimated as FPKM with Cufflinks v2.0.0 (Trapnell et al. 2010). The procedure was repeated for unambiguously mapped reads only and for all mapped reads, using multiread and fragment-bias correction methods as implemented in Cufflinks where applicable. We used these two estimates of expression levels to identify gene copies for which expression levels can be determined with certainty and flag those for which reliable expression values cannot be estimated (Supplemental Methods). We checked our method against a data set of “problematic” human genes, for which expression levels cannot be reliably estimated (Robert and Watson 2015). Our approach of filtering and flagging effectively removes problematic genes (Supplemental Methods). Importantly, flagged genes and gene families in all species were excluded from any analysis that required gene-specific expression levels. The final expression levels were calculated for each gene using all reads (even if no unique reads were present) and employing the –multiread-correct option.
Because expression profiles of the two neural tissues, cortex and cerebellum, are highly correlated, we computed the mean of their expression for each gene and used this value in all subsequent analyses. We validated the results treating brain and cerebellum as separate tissues (Supplemental Methods). We normalized the expression levels among samples with a median scaling procedure (Brawand et al. 2011) and calculated species median expression levels for each tissue. All expression levels were log2-transformed. To be able to take the logarithm of all values, we set the smallest value to 10−6 and replaced all values smaller than 10−6 with it.
Statistical analysis
All statistical analyses were performed in R 2.15.3 (R Core Team 2012). Significance levels were adjusted with Benjamini–Hochberg correction (Benjamini and Hochberg 1995). Significant χ2 tests were followed by a post hoc procedure as implemented in the R package polytomous (https://artax.karlin.mff.cuni.cz/r-help/library/polytomous/html/00Index.html). We used standardized Pearson residuals to assess if individual observed values differ significantly from an overall hypothetical homogeneous distribution and to identify the direction of these differences (over- or underrepresentation) by tissue or by duplication age. A Wilcoxon rank-sum test was used to study differences in expression divergence between species-specific paralogs and 1:1 orthologs.
To evaluate the contribution of different factors (expression levels, duplication status [i.e., paralog or single-copy gene], evolutionary time [duplication or speciation age]) to expression divergence and tissue specificity, we constructed linear models in R that included all orthologous relationships between human and other species as well human paralogs of different duplication ages. Human was chosen as, given our collection of species, it provides the most detailed data on duplication and speciation ages. Qualitatively similar results were obtained with other focal species. Because most paralogs in our data set have much older duplication ages than the oldest speciation age of the single-copy genes (human-chicken divergence) and this can affect the linear model, we repeated the analyses by removing all paralogs with duplication ages older than amniotes. We also repeatedly (100 times) subsampled as many single-copy genes as there are paralogs to match the sample sizes of both gene types.
Gene Ontology analysis
Overrepresentation of Gene Ontology terms (The Gene Ontology Consortium 2000) in human and mouse genomes were identified in GOrilla (Eden et al. 2009). This tool allows either finding enrichments in a ranked gene list or evaluating functional overrepresentation in a candidate data set against a specified list of background genes. We set the false discovery rate (FDR) of 0.1% as our cutoff value and employed the Benjamini–Hochberg correction for multiple testing within each data set.
Expression divergence and protein sequence divergence
Expression divergence was calculated by species for normalized, log-transformed expression values across all available tissues in a pairwise manner across sister clades resulting from a given duplication event (Supplemental Methods). Protein sequence divergence (dN) was calculated on the basis of pairwise alignments between paralogs using the longest transcript of each paralog. Spearman's correlation coefficients were calculated between expression divergence and protein sequence divergence for human and opossum paralogs by duplication age. To avoid the potential bias introduced by a small number of highly expanded gene families, we randomly sampled a single gene pair per gene family and calculated the correlation between expression and protein sequence divergence on this data set.
To assess if genes that differ in their extent of protein sequence divergence compared to expression divergence and vice versa fulfill different biological functions, we ranked paralog pairs in two different ways, following Warnefors and Kaessmann (2013): (1) genes with higher expression divergence rank relative to their protein sequence divergence rank were classified as ED-biased; and (2) genes with higher protein sequence divergence rank relative to their expression divergence rank were classified as dN-biased. We also grouped genes into two age classes: lineage-specific and young paralogs that have duplicated in the common amniote ancestor or more recently; and old and ancient paralogs that have duplicated in the tetrapod ancestor and earlier (see below for more details). We performed GO enrichment analyses separately for these groups as described above.
Duplication age groups
For our analyses of evolutionary dynamics of tissue specificity we defined four groups of duplication ages: (1) lineage-specific paralogs, e.g., primate-specific, rodent-specific, marsupial-specific, etc.; (2) “young” paralogs that duplicated along the branch leading to the amniote ancestor and are older than lineage-specific duplications; (3) “old” paralogs with duplication ages in the tetrapod and bony vertebrate (Euteleostomi) ancestors; and (4) “ancient” paralogs with duplication ages in the ancestors of vertebrates, chordates, coelomates, and bilaterals. For the analysis of placenta-specific expressed paralogs, we subdivided the “young” category into genes that emerged in the common eutherian ancestor or before that, on the branch leading to the ancestor of therians and amniotes.
Tissue specificity
We used two measures of tissue specificity: tau (Yanai et al. 2005), and relative gene expression. For the second measure, we defined tissue specificity as relative expression of the gene in the tissue with highest expression (expMaxTissue/sum[expAllTissues]). This calculation was performed on normalized, but not transformed, expression values. For a set of n tissues, uniformly expressed genes show tissue specificity of 1/n, whereas genes expressed in a single tissue show tissue specificity of 1, independent of n. Relative tissue expression was calculated as the expression of a gene in the target tissue divided by the sum of its expression in all tissues. Both measures of tissue specificity (tau and relative gene expression) were strongly correlated with each other in our data set (rho = 0.968, P < 10−15).
Genes with tau ≥ 0.8 were defined as tissue-specific expressed. Using relative gene expression, tissue-specific expressed genes were defined in two different ways. For most of our analyses, genes were considered tissue-specific expressed if they showed at least twofold higher expression in the tissue of highest expression than under uniform expectation: 2/n (“twice uniform expression”). In the alternative, more stringent definition of tissue specificity, we required the tissue-specific expressed gene to show at least twofold higher expression in tissue with highest expression than in any other tissue (“stringent”). To evaluate the correlation between expression levels and tissue specificity, we assessed expression levels as means across all tissues.
To test for changes in the proportion of genes with the highest expression in a given tissue, we calculated for each tissue and in each species the number of genes with the highest expression in this tissue for each age category (as shown in Fig. 4C). We then applied a χ2 test, followed by a post hoc procedure (as described above) to identify age categories with significantly more or fewer genes.
Lineage-specific expansions of amniote gene families
Only gene families with at least three paralogs that emerged in the common amniote ancestor or more recently for any given species were considered in this analysis, as we were interested in studying how repeated duplications influence the lineage-specific repertoire of paralogs. We used twice uniform expression (see the previous section) to define tissue-specific expressed genes, and only retained paralogs for which gene-specific expression patterns could be unambiguously inferred. We also evaluated duplications along individual amniote lineages that were not present in their common ancestor (human paralogs that have duplicated along the primate lineage, mouse paralogs that have duplicated along the rodent lineage, etc.) with the aim of identifying gene families that have repeatedly and independently produced lineage-specific expansions (Fig. 7; Supplemental Table S10). We treated them in the same way as above, requesting at least three paralogs per species for which expression patterns could be unambiguously determined. Including genes for which the expression patterns could be confounded by closely related paralogs did not change the results (Supplemental Table S14).
Acknowledgments
This work was supported by grants from the European Research Council (Starting Grant: 242597, SexGenTransEvolution) and the Swiss National Science Foundation (Grant: 130287) to H.K., by the European Molecular Biology Organization long-term fellowships EMBO ALTF 1051-2010 to K.G. and EMBO ALTF 1589-2011 to M.W., and the Human Frontiers Science Program long-term fellowship (HFSP: LT000800/2011) to K.G. We thank Julien Meunier, Magali Soumillon, Francesco Carelli, Philippe Julien, Adem Bilican, Anamaria Necsulea, Diego Cortez, and other members of the H.K. group for support and discussions throughout the project, Tomas Marques-Bonet and Margarida Cardoso Moreira for helpful comments on the manuscript, and three anonymous reviewers for insightful and helpful comments on this work.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.215566.116.
- Received September 5, 2016.
- Accepted July 18, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








