

Abstract
Advances in the development of high-throughput screening and automated chemistry have rapidly accelerated the production of chemical and biological data, much of them freely accessible through literature aggregator services such as ChEMBL and PubChem. Here we explore how to use this comprehensive mapping of chemical biology space to support the development of large-scale quantitative structure-activity relationships (QSAR) models. We propose a new Deep Learning Consensus Architecture (DLCA) that combines consensus and multitask deep learning approaches together to generate large-scale QSAR models. This method improves knowledge transfer across different target/assays while also integrating contributions from models based on different descriptors. The proposed approach was validated and compared with Protochemometrics, Multitask Deep Learning and Random Forest methods paired with various descriptors types. DLCA models demonstrated improved prediction accuracy for both regression and classification tasks. The best models together with their modeling sets are provided through publicly-available web services at https://predictor.ncats.io.
INTRODUCTION
During the last decade, the amount of publicly-available chemical and biological data has rapidly grown, supported by advancements and the availability of high-throughput screening approaches.1,2 There are now several web sites, including ChEMBL3 and PubChem,4 that aggregate data from published screening and medicinal chemistry efforts. Indeed, the number of compounds published in ChEMBL between 2010 and 2017 has increased 3 times and the number of biological assays or end-points for these compounds has increased 5.9 times. This volume of data should enable the comprehensive mapping of chemical-biology space revealing interrelated relationships between chemical compounds, biological targets and diseases and accelerate the development of large-scale quantitative structure-activity relationships (QSAR) models. Several research studies have attempted to construct such large-scale models. Koutsoukas et al.5 created QSAR models based on the extended connectivity fingerprints and Laplacian-modified Naive Bayes classifier and Parzen-Rosenblatt Window using data from ChEMBL (ver 10). Pogodin et al.6 used data extracted from the ChEMBL (ver. 19) database to construct Bayesian-like model implemented in PASS software for the prediction of interactions between 2507 protein targets and drug-like compounds. Clark et al.7 used extended connectivity (ECFP) and molecular function class (FCFP) fingerprints together with Laplacian-corrected naive Bayesian for the construction of 2000 classification models based on ChEMBL (ver. 20) data. None of these models share information between targets during model building. Indeed, these conventional QSAR methods ignore how targets relate to one another, how insights from one model system might be transferred to another and thus provide a limited capacity for extrapolation to undercharacterized targets. Recently, Varnek et al. showed8 that aggregated results from the different related biological targets could further improve the quality of QSAR models. There are several approaches which are capable of performing inductive transfer across different related targets including multitask deep learning (MDL) and proteochemometrics (PCM). Ramsundar et al.,9 applied a multitask neural network for modeling of 259 datasets gathered from publicly available data including 128 bioassays from PubChem and showed that MDL was superior to single task models (conventional QSAR models). Kearnes et al.,10 have also applied MDL to the set of 22 ADMET datasets from Vertex including hERG inhibition, aqueous solubility, compound metabolism, and others. They revealed that while MDL performed better than traditional modeling of each dataset independently, the differences in performance were marginal. Mayr et al.,11 have investigated binary classification problems using ChEMBL data and compared MDL with conventional machine learning approaches together with graph convolutional and SMILES based LSTM models. Authors revealed that fingerprints and descriptors based MDL models outperformed all other models. However, authors have not studied regression problems as well as consensus modeling. Further investigations of MDL confirmed12 its advantage over traditional methods and explained13 the nature of these improvements. Thus, the usage of chemogenomics data paired with deep neural nets may lead to improvement of large-scale predictions.
Another approach which implements inductive transfer is proteochemometrics (PCM).14 This approach uses descriptors of chemical compounds together with descriptors of proteins. This allows PCM models to make predictions even for new targets. Several researches15,16 have shown that PCM models perform better than classical QSAR models. In fact, Cheng et al.,17 have shown that if QSAR is extended to multitarget predictions it can outperform PCM modeling. Recently, Lenselink et al.,18 compared the performance of MDL and PCM based on deep learning models for binary classification problems using ChEMBL data (ver. 20). Both approaches yielded similar results with a slight advantage for PCM deep learning models. In these detailed methods comparisons, the authors focused only on classification problems and did not investigate applying these methods to regression tasks. Also, the authors arbitrarily selected a compound activity cut-off for classification of 300 nM, to help artificially balance the data sets in roughly equal partitions (55/45%). Thus, the obtained artificially-balanced validation sets do not resemble more common distributions of imbalanced data19 that screening campaigns typically produce. A fairer validation would explore modeling performance on more naturally-distributed imbalanced screening results.
Additional improvements in modeling performance can be achieved by applying consensus modeling approaches. Consensus models combine outputs from several models created using different sets of descriptors and/or different machine learning techniques. Predictions from the models can be arithmetically averaged (simple unweighted consensus) or can be averaged with some weights for each model (weighted consensus). It has been shown20,21 that consensus models have a reduced variability compared to individual models, which leads to more reliable and accurate predictions. Indeed, different descriptors have different strengths and weaknesses and allow one to look at the classification problem from different angles. Aggregation of these models into a consensus model allows one to achieve the improvement in the model’s performance. Consensus modeling is being broadly used for development of conventional QSAR models, and is beginning to be adopted22 in the creation of deep learning models. However, there are no publicly available, easy to use, consensus deep learning models created using large-scale chemogenomics data. Some aggregation of models outputs can be made by employing a stacking approach, where one classifier is stacked on top another one. For instance, Martin et al.,23 implemented stacked QSAR modeling in the so-called profile-QSAR approach. This method is based on Morgan fingerprints from RDkit package with five physical-chemical properties (ALogP, MW, number of H-bond acceptors, number of donors, and number of rotatable bonds) combined with stacking of two machine learning approaches: RandomForest and PLS, using publicly available data of 171 kinase assays from PubChem, and 159 kinase assays from ChEMBL, and internal data of 728 Novartis kinase assays. One limitation of this approach is that their profile-QSAR approach employs predicted data, which includes the combined errors from experimental and modeling parts. The usage of MDL approach in this case might improve the quality of the models since it does not rely on putative data during utilization of the transfer knowledge across different targets.
Summarizing the studies mentioned above, the combination of MDL or PCM approaches with consensus modeling may provide an opportunity to boost the optimal performance of QSAR models. However, it is unclear which combined methods would produce the best performance overall. In particular, a weighted consensus approach requires the utilization of corresponding weighting parameters, and the selection of these are not obvious. We think that the flexibility of deep learning techniques opens up a greater opportunity to incorporate consensus models inside and allows it to learn the best consensus model during back propagation. In this work, we propose a new deep learning architecture, in which we incorporate the consensus approach of multitask models based on different descriptors and compare this approach with regular consensus models of MDL and PCM methods using the same descriptors sets.
MATERIALS AND METHODS
DATA SETS
To properly validate the proposed MDL and PCM approaches we collected and compiled datasets for both regression and classification problems using two publicly available resources: ChEMBL and Tox21 databases.
ChEMBL modeling set
ChEMBL is a publicly-available database of bioactive drug-like small molecules, containing 2-D structures, calculated properties (e.g. LogP, Molecular Weight, Lipinski Parameters, etc.) and bioactivities extracted from literature (e.g. binding constants, pharmacology and ADMET data). This database was developed and is maintained by the European Bioinformatics Institute (EMBL-EBI). In this study we used ChEMBL version 23, which includes sparse bioactivity information about 1,735,442 unique compounds against 11,538 targets. From ChEMBL database (ver. 23) we selected 1082 Homo sapiens protein targets (from 4,108 available), which have at least 10 compounds with IC50 values. We eliminated all compounds with inconclusive results and kept only compounds which obeyed the following: i) assay confidence score more than 6, ii) operator of activity equals to “=“, iii) bioactivity expressed in “IC50” values, iv) activity unit is “nM”. All IC50 values were converted in pIC50 = -Log(IC50,[M]). Multiple measurements for the same compound and target were averaged if their standard deviation was less than 0.5 logarithmic units, otherwise they were discarded. This produced a dataset of 354,000 bioactivity values for 251,998 compounds against 1082 targets (Table S1.1). Thus, ChEMBL modeling data are extremely sparse (Figure 1A), providing measured values for only 0.13% of the potential compound-target matrix.
Figure 1.
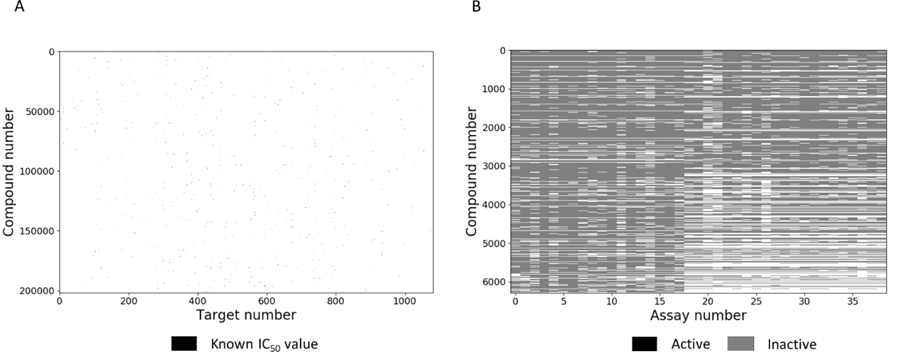
Activity matrix of (A) ChEMBL and (B) Tox21 data sets.
Tox21 modeling set
To generate a dataset for classification modeling we used data gathered from toxicity-related assays screened through the Tox21 initiative.24 The Toxicology in the 21st Century (Tox21) program is a federal collaboration among NIH’s NCATS and the National Toxicology Program at the National Institute of Environmental Health Sciences; the Environmental Protection Agency; and the Food and Drug Administration. Tox21 initiative aim to develop better toxicity assessment methods to quickly and efficiently test whether certain chemical compounds have the potential to disrupt processes in the human body that may lead to negative health effects.
From Tox21 database25 we selected 39 assays (Table S2.1) which have at least 10 active compounds. The selected assays represent different nuclear receptors signaling and stress pathways. From 39 assays 38 are target specific and one is phenotypic assay (screening small molecules which disrupt the mitochondrial membrane potential). All compounds were screened in triplicates. We eliminated all inconclusive data and kept only active and inactive compounds using an activity threshold of 10 µM. The final dataset includes 7857 compounds and their activity values across all 39 assays. Only a small amount of compounds (less than 5%) were considered as inconclusive for particular assays. Overall the activity matrix (Figure 1B) consists of 306,423 experiments results (active, inactive and inconclusive) with 11,915 active ones (3.8%).
METHODS
Descriptors
To develop MDL and PCM models we calculated three different types of fingerprints as chemical descriptors using RDkit software and PROFEAT descriptors as target-based descriptors.
RDkit software package26 was used to calculate i) Morgan fingerprints, ii) Avalon fingerprints and iii) AtomPair fingerprints. These types of fingerprints were selected for their diversity of approaches and their capture of different functional groups and features. Indeed, Morgan fingerprints are circular-based fingerprints commonly used for drug discovery7 and similarity purposes.27 Avalon fingerprints28 are path-based fingerprints which take into account not only structure fragments, but also different structure features (number of bonds contained in rings of different sizes, graph distance pairs for special end point atom types, etc.). AtomPair fingerprints29 are path-based fingerprints encoding pairs of atoms together with the number of bonds separating them and are often used for substructures searching.30 All fingerprints were calculated with length of 1024 bits and for Morgan fingerprints a radius of 2 was used.
The PROFEAT web server31 was used to calculate descriptors for protein targets. The service allows one to calculate 14 different classes of descriptors and proteins features from amino acid sequence (Amino acid composition, dipeptide composition, autocorrelation descriptors, etc.). In total, 1437 descriptors were calculated for each target sequence. All PROFEAT descriptors were normalized using min-max normalization function, which transforms all values in the range between 0 and 1.
Machine learning approaches
For model development we used three machine learning approaches: i) RandomForest, ii) Deep neural net and iii) a new, customized learning architecture.
Random Forest
Random Forest (RF) models were obtained using scikit-learn32 package implemented in python. RF is an ensemble of the decision trees. More trees reduce the variance. The classification from each tree can be thought of as a vote; the most votes determine the classification. The regression output is calculated as a mean value of all trees. Each tree has been grown as the following. A random sample of compounds (67%) is selected from the initial modeling set as the training set for the current tree. Not selected samples are using as a test set called an out-of-bag (OOB), which typically is 33% of initial modeling data. The randomly selected descriptors from the training set are used to split the nodes in the tree. Each tree is grown until it reaches the maximum tree depth parameter. The internal model evaluation has been done according to the performance on the OOB set. The number of trees used in this study was 500.
Totally, we built three conventional RF models based on each type of fingerprints (Morgan, Avalon, AtomPair) and three proteochemometrics models (PCM_RF) based on combination of each type of fingerprints with PROFEAT descriptors.
Deep neural net
In this study, we used the multi-layer feedforward neural networks implemented in Keras33 using the Tensorflow34 backend. For minimization of the loss function we used the ADAM algorithm,35 which computes individual adaptive learning rates for different parameters from estimates of first and second moments of the gradients. Since there are plenty of parameters which need to be selected or optimized to construct the predictive neural net model, we used a grid search technique to reveal the best hyperparameters. Thus, the grid search was performed across the following parameters: i) number of hidden layers {2, 3, 4}, ii) number of neurons {8000, 6000, 4000, 3000, 2000, 1000, 700, 500}, iii) learning rate {0.01, 0.001, 0.0001}, iv) activation function {ReLU, tanh}, v) batch size {32, 128, 256, 512}, and vi) number of epoch {30, 70, 100, 150, 200}. Grid search was performed over “tanh” and “ReLU” functions, as successfully validated in several recent computational drug discovery studies.36,37,38,18 “SELU” was recently reported39 to provide superior over “ReLU” function, especially for cases with more than 8 hidden layers such as Highway, ResNet, etc. As we were limited by size of hidden layers of 4 due to computationally expensive calculations, we have not used “SELU” approach during hyperparameter optimization. In this study, we built both single- and multi-task models. The difference between single- and multi-task models was the number of outputs. Thus, single-task model has one output node with linear or sigmoid activation function for regression or classification problem. Multi-task model has multiple output nodes predicting targets/assays endpoints with corresponding linear or sigmoid activation.
In total, we built three multi-task deep learning models (MDL) for each type of fingerprints and three proteochemometrics deep learning models (PCM_DL) based on single-task architecture and combination of each type of fingerprints with PROFEAT descriptors.
Concatenation of descriptors
In addition to separate MDL models, we also built a MDL model based on the concatenation of fingerprints. We named it MDL_concat, and this model followed the same parameter optimization procedures as the separate MDL models.
Consensus modeling
The consensus model for each approach (RF and MDL) was developed by averaging the prediction results obtained from three models based on each type of fingerprints (Morgan, Avalon, AtomPair) with combination of PROFEAT descriptors for proteochemometrics approaches (PCM_RF, PCM_DL). Thus, four consensus models were constructed: i) consensus Random Forest model (Consensus_RF), ii) consensus multi-task deep learning model (Consensus_MDL), iii) consensus Random Forest for proteochemometrics (Consensus_PCM_RF) and iv) consensus deep learning model for proteochemometrics (Consensus_PCM_DL)
New deep learning consensus architecture
Here we also propose a new deep learning architecture which incorporates a consensus approach inside the neural network. Thus, we combine three separate MDL networks built based on three types of fingerprints by averaging their outputs (see Figure 2) inside the single neural net, and therefore, forcing the learning algorithm to propagate the corresponding errors and improving the consensus results. In this case, during learning, the neural net is figuring out the best weights for each particular output as well as for their averaging. As a result, the network provides multi-task outputs for each type of fingerprint, as well as a consensus output for all multi-task outputs. We name this approach DLCA (deep learning consensus architecture).
Figure 2.
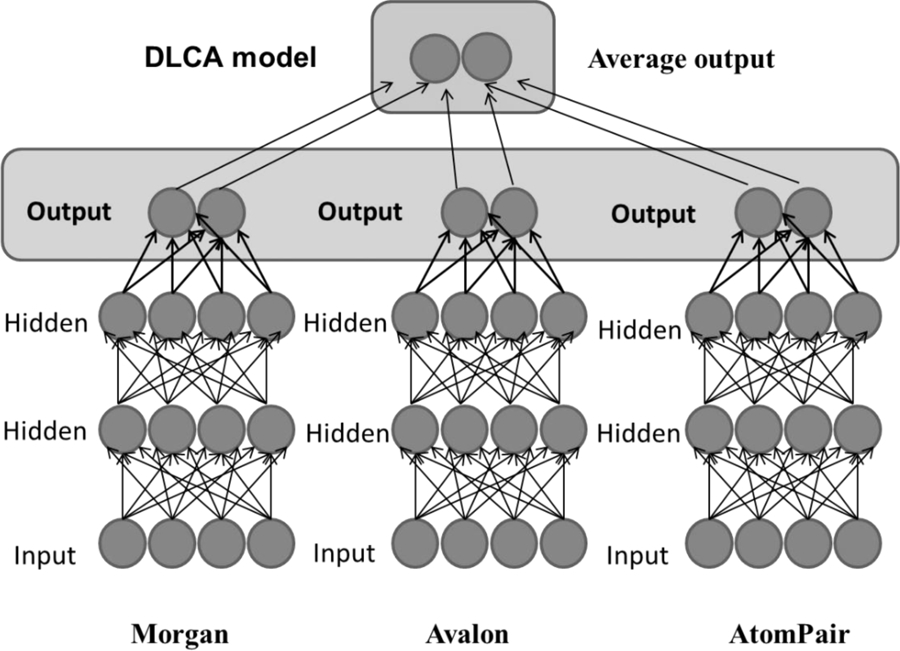
Proposed new deep learning consensus architecture.
Validation procedure
To validate the developed models, we simulated the typical drug discovery case in which an available data set from a screening campaign is used to build a model and then it is applied to another chemical library for compound prioritization. Thus, the prepared modeling sets were randomly divided into training and test sets using 80% and 20% of the data respectively. To select the best hyperparameters for deep learning models we used a 5-fold cross-validation procedure (5-fold CV) utilizing only the training set data. During this procedure, the initial training set was randomly subdivided into 5 parts. Four parts were used as the internal training set for model building and remaining part was used as the internal test set for the assessment of predictive accuracy. The 5-fold CV procedure was repeated many times during grid search until the best hyperparameters were revealed. After hyperparameters were selected the model was rebuilt using the entire training set and resulting model was applied to our hold-out test set of data the model had never seen.
Evaluation of the model prediction accuracy
For estimating the prediction accuracy, the following statistical parameters were calculated.
For classification models:
1) Sensitivity: accuracy of predicting “positive” (active) when the true outcome is positive.
where TP: true positive and FN: false negative.
2) Specificity: accuracy of predicting “negative” (inactive) when true outcome is negative.
where TN: true negative and FP: false positive.
3) Balanced Accuracy: Average between Sensitivity and Specificity.
4) AUC (area under the receiver operating characteristic curve): it is a graph showing the performance of a classification model at all classification thresholds.40
This curve plots two parameters: i) Sensitivity and ii) 1-Specificity.
For regression models:
5) Squared Pearson’s coefficient
Where y is observed value for each particular compound, x is predicted value for each particular compound, is average observed value, is average predicted value and n is the number of objects in the training set.
6) Root mean square error
Where is predicted value for each particular compound, Yi is observed value for each particular compound and n is the number of objects in the training set.
RESULTS
Development and comparison of baseline models
Regression task with ChEMBL modeling set
ChEMBL modeling data was divided onto training and test set (cf. Methods) resulting in 201,599 compounds included in the training set and 50,399 compounds included in the test set. For each fingerprints type (Morgan, Avalon, AtomPair), we constructed RF and MDL models. In addition, we also built and MDL_concat model based on descriptors concatenation. Using a 5-fold CV procedure, the optimal hyperparameters for each MDL model were established resulting in the following values: i) number of hidden layers {4}, ii) number of neurons for each layer {4000, 2000, 700, 500}, iii) learning rate {0.0001}, iv) activation function {ReLU}, v) batch size {32}, and vi) number of epoch {200}. The protochemometrics (PCM) models for each combination of fingerprints and PROFEAT descriptors (Morgan and PROFEAT, Avalon and PROFEAT, AtomPair and PROFEAT) were constructed using RF and DL as well. The best parameters for PCM-DL models were found: i) number of hidden layers {4}, ii) number of neurons for each layer {8000, 4000, 700, 500}, iii) learning rate {0.0001}, iv) activation function {ReLU}, v) batch size {128}, and vi) number of epochs {100}. Once parameters were established the deep learning models were rebuilt using the entire training set. In total, 13 models were developed, which are represented by three models per approach and one model based on descriptors concatenation (MDL, RF, PCM_DL, PCM_RF and MDL_concat). The developed models were applied to the test set and performances of each model were calculated. To avoid outliers in statistical analysis the models predictivity were estimated only for those targets which have ten or more compounds in the test set, resulting in 820 targets from 1082. Statistical characteristics of these models for all 820 targets can be found in the supplementary material (Table S1.1, S1.2). Average prediction results calculated using RMSE and R2 across all targets for each model are presented in Figure 3.
Figure 3.
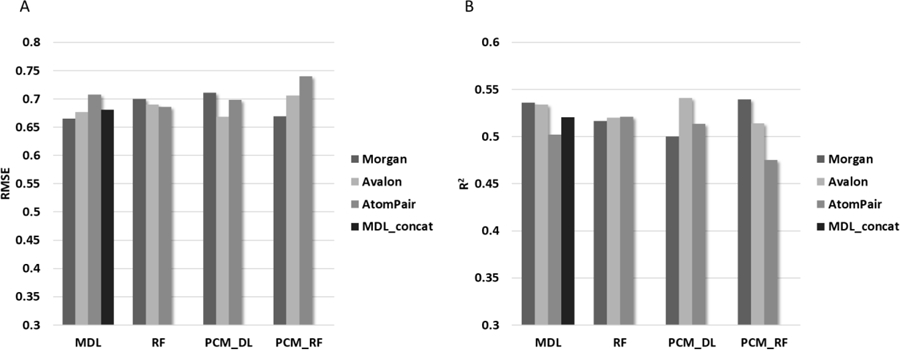
(A) Overall RMSE and (B) R2 of prediction for the sets of MDL, RF, PCM_DL, PCM_RF and MDL_concat regression models using data taken from ChEMBL.
(A) Average root mean square error (RMSE) values calculated across all targets for external test set. (B) Average R2 values calculated across all targets for external test set.
Both RMSE and R2 have a good correlation across 13 models. Indeed, the ranking of the height of bars on Figure 3A are almost the complete opposite of that in Figure 3B. Figure 3A and B show that the best prediction results are obtained with Morgan fingerprints and the MDL approach. The second-best results were achieved using Avalon fingerprints with PCM_DL and Morgan fingerprints with PCM_RF. It is interesting to note that each type of fingerprint performs differently depending on machine learning approach employed. Thus, there is not one dominant type of fingerprints across all approaches. Indeed, Morgan fingerprints shown better performance with MDL and poorer performance with PCM_DL. Avalon fingerprints show good performance with PCM_DL but poor accuracy with PCM_RF. The best results for AtomPair fingerprints are obtained with RF and the worse ones are found with PCM_RF. Concatenation of descriptors have not improved the model performance compared to the separate models. In fact, the obtained results from MDL_concat model were mostly centered between the best and worse results of the separate models. In general, each type of fingerprints works best only in combination with certain types of machine learning approaches. However, the utilization of consensus modeling, as shown previously,20,21 can further reduce the variance across different fingerprints and methods.
Classification task with Tox21 modeling set
Tox21 modeling data was divided into training and test set (cf. Methods) resulting in 6,286 compounds in the training set and 1,571 compounds in the test set. Since this is an internal library of compounds which we screened under the Tox21 initiative, all prediction results obtained for compounds in the test set can be considered as a prospective validation. For each fingerprints type (Morgan, Avalon, AtomPair), we constructed RF, PCM_RF, PCM_DL and MDL models. However, PCM_RF and PCM_DL were developed using PROFEAT descriptors as well. In addition, we also built a MDL_concat model based on concatenation of fingerprints. Hyperparameters for the MDL model were found to be optimal with the following values: i) number of hidden layers {4}, ii) number of neurons for each layer {4000, 2000, 1000, 700, 500}, iii) learning rate {0.0001}, iv) activation function {ReLU}, v) batch size {128}, and vi) number of epochs {30}. The best parameters for PCM-DL models were found: i) number of hidden layers {4}, ii) number of neurons for each layer {6000, 3000, 700, 500}, iii) learning rate {0.0001}, iv) activation function {ReLU}, v) batch size {128}, and vi) number of epochs {100}. It is interesting to emphasize that the number of neurons for PCM models is higher compared to the MDL model, which can be explained by the fact what PCM models have a larger number of training samples. Indeed, the number of training samples for MDL model corresponds to the number of compounds; however, the number of samples for PCM corresponds to the number of compounds multiplied by the number of targets. In total, 13 models were developed. In contrast to the ChEMBL modeling exercise, most compounds from the Tox21 set were screened across all assays and thus, the predictivity of models were calculated for all 39 assays. Statistical characteristics of these models can be found in the supplementary material (Table S2.1, S2.2). Average prediction results calculated using AUC and balanced accuracy across all assays for each model are presented in Figure 4.
Figure 4.
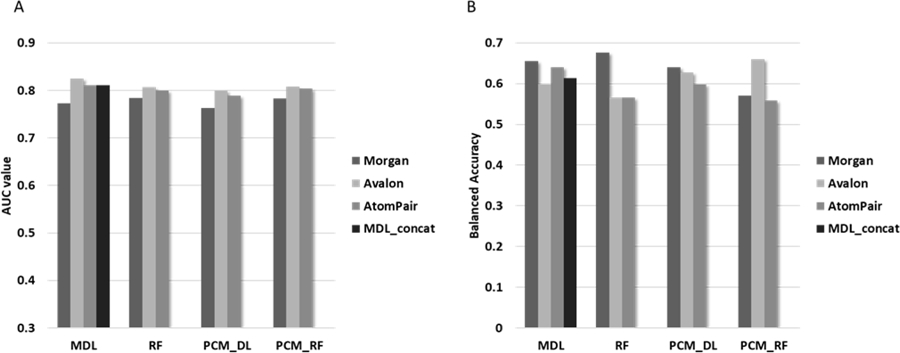
Accuracy of prediction for the 13 classification models based on Tox21 data set.
(A) Average AUC values calculated across all assays for external test set. (B) Average balanced accuracy values calculated across all assays for external test set.
In the case of classification models results, there is no clear correlation between AUC and balanced accuracy (BA) values. Indeed, it can be seen from Figure 4 that the best results in terms of AUC were achieved with Avalon fingerprints and MDL. However, according to BA values, the best results were found for Morgan fingerprints and RF. From an AUC performance perspective, the Avalon fingerprints outperformed all other fingerprints. Morgan fingerprints performed worse according to AUC results. In contrast to that, the BA results showed an absence of any particular fingerprint method being superior. Indeed, considering all fingerprints models together the MDL and PCM_DL methods on average performed better, but individually RF and PCM_RF showed better balanced accuracy results. Similar results were found in Tox21 Data Challenge41,42 organized by NCATS in 2014.43 The different methods showed better performances across twelve different Tox21 assays. It is necessary to emphasize that the concatenation of descriptors has not improved the prediction results compared to separate ones. However, as we mentioned above, a consensus approach can further reduce variance and may offer an improved approach.
Development and comparison of consensus models
Consensus models driven from ChEMBL modeling set
From the 12 regression models described above we constructed 4 consensus models, one per method, by averaging the prediction results from each fingerprints model. Thus, Consensus_MDL, Consensus_RF, Consensus_PCM_DL and Consensus_PCM_RF were developed. In addition to these classical consensus models we propose (cf. Method) a new deep learning architecture which incorporates consensus modeling inside of the neural net. We name this approach DLCA (deep learning consensus architecture). As with other individual models, the 5-Fold CV procedure was used to established hyperparameters for the DLCA model. The best accuracy was found with the following values: i) number of hidden layers {4}, ii) number of neurons for each layer {6000, 3000, 700, 500}, iii) learning rate {0.0001}, iv) activation function {ReLU}, v) batch size {128}, and vi) number of epochs {150}. Once the hyperparameters were established, we rebuilt DLCA model using entire ChEMBL training set and applied it to the test set. The comparative analysis of the prediction results obtained from 5 consensus models are presented in Table 1.
Table 1.
Accuracy of prediction for the 5 consensus models based on ChEMBL data set.
| Average RMSE value | Average R2 value | |
|---|---|---|
| Consensus_MDL | 0.651 | 0.565 |
| Consensus_RF | 0.649 | 0.567 |
| DLCA_model | 0.637 | 0.579 |
| Consensus_PCM_DL | 0.645 | 0.567 |
| Consensus_PCM_RF | 0.68 | 0.545 |
Average root mean square error (RMSE) values calculated across all targets for external test set. Average R2 values calculated across all targets for external test set. The best results are shown in bold.
Table 1 shows that DLCA produced superior RMSE and R2 values. The differences between DLCA and other consensus models results are statistical significant according to Wilcoxon matched-pairs signed rank test with p value <0.0001. Both RMSE and R2 results have a good correlation across the consensus models. The second-best results were obtained by the Consensus_PCM_DL model and the worst results by the Consensus_PCM_RF method. It is interesting to emphasize that comparing results in Figure 3 and Table 1, one might see that all consensus models outperformed the corresponding individual ones. However, comparing the methods together it can be seen that some surprising results were observed. Indeed, two out of three individual MDL models were better compared to RF ones (Figure 3), though the Consensus_RF showed a slightly better accuracy than Consensus_MDL. Although it was previously shown9,38 that in general, individual deep learning models outperform Random Forest ones, it might not be the always case with consensus modeling. It is interesting to further compare the performances of the different approaches considering the number of targets producing acceptable models. We calculated the number of targets which passed the certain cut-off for each approach. The following criteria were used as the thresholds for these calculations: i) R2 >= 0.6 which was recommended elsewhere44 and ii) RMSE <= 0.65, which was obtained as average RMSE across all consensus methods. The obtained results are presented in Figure 5.
Figure 5.
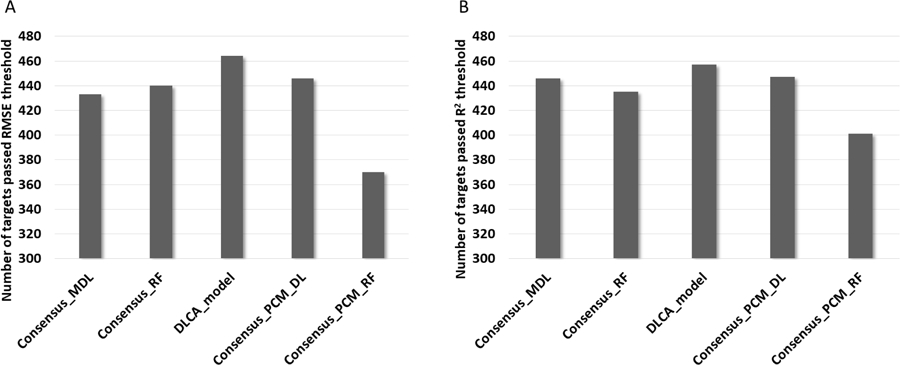
Target applicability of 5 consensus models based on ChEMBL data set.
(A) Number of targets predicted with RMSE <= 0.65 calculated for external test set. (B) Number of targets predicted with R2 >= 0.6 calculated for external test set.
According to the results in Figure 5, four methods out of five produced acceptable predictions for more than half of targets using these selected thresholds for the both RMSE and R2 values. The DLCA model showed the best performance being ahead of Consensus_PCM_DL on 18 and 10 targets for RMSE and R2 thresholds, respectively. It is interesting to notice that results obtained for Consensus_MDL and Consensus_RF are anti-correlated. Indeed, Consensus_MDL is 7 targets behind of Consensus_RF for RMSE values and 11 targets ahead for R2 values. This observation reveals the importance of analysis which not only calculates the average of the prediction results across of all targets, but also counts the number of predictive targets obtained from each model. Certainly, in some cases, it might be better to have a larger coverage across predictive targets rather than having the better average accuracy, since it considers the poor predictive targets as well. The worst results are found using the Consensus_PCM_RF method, which can be explained by contribution of the poorly performing individual models based on AtomPair fingerprints.
Consensus models driven from Tox21 modeling set
Similar to the ChEMBL task, we constructed 4 consensus classification models from 12 individual ones, one per method, by averaging the prediction results from each fingerprints model. In addition, we developed a DLCA Tox21 model considering hyperparameter optimization procedure during 5-Fold CV. The optimal parameters for this model were found: i) number of hidden layers {4}, ii) number of neurons for each layer {2000, 1000, 700, 500}, iii) learning rate {0.0001}, iv) activation function {ReLU}, v) batch size {32}, and vi) number of epochs {70}. The selected parameters were used to construct final DLCA model using the entire Tox21 training set. After that all 5 consensus models were applied to the test set. The average prediction performances of the methods are presented in Table 2.
Table 2.
Accuracy of prediction for the developed 5 consensus models based on Tox21 data set.
| Average AUC value | Average BA value | |
|---|---|---|
| Consensus_MDL | 0.833 | 0.658 |
| Consensus_RF | 0.832 | 0.573 |
| DLCA_model | 0.84 | 0.673 |
| Consensus_PCM_DL | 0.817 | 0.621 |
| Consensus_PCM_RF | 0.828 | 0.566 |
Average AUC values calculated across all assays for external test set. Average balanced accuracy values calculated across all assays for external test set. The best results are shown in bold.
Comparing the results in Table 2 and Figure 4 it can be seen that all consensus models outperformed the individual ones using the AUC metric. However, in terms of balanced accuracy the Random Forest based consensus models did not show an improvement compared to individual ones. The large performance difference between the best model and remaining ones is the reason for that. Indeed, two out of three models were worse than the best one for both RF and PCM_RF modeling approaches (Figure 4). The averaging of results from the three models pulls down the overall consensus performance to level of the worst models. An alternative solution for this problem might be a weighted consensus technique in which the models outputs are weighted to the some parameters, such as the models performance values,45 the applicability domain,46 etc. In this study the weighted consensus did not improve (not shown) the prediction results.
Identical to the ChEMBL modeling results, the DLCA model showed the best accuracy of prediction in terms of both AUC and BA values. It is not surprising, since DLCA architecture is using deep neural net to figure out the contribution of each internal model and thus, it establishes the best non-linear way to weight each consensus output. Second-best results were achieved by Consensus_MDL approach leaving the remaining ones with ambiguous performances. Thus, Consensus_RF and Consensus_PCM_RF showed the good AUC values, but at the same time the poor BA results. The opposite observation was found for Consensus_PCM_DL.
Similar to ChEMBL consensus model’s comparison, we calculated the number of assays which passed the certain performance cut-offs for each approach. The following criteria were used as the thresholds for these calculations: i) AUC >= 0.85, which is close to average AUC value (0.83) and ii) BA >= 0.65, which is close to average BA value (0.62). The obtained results are presented in Figure 6.
Figure 6.
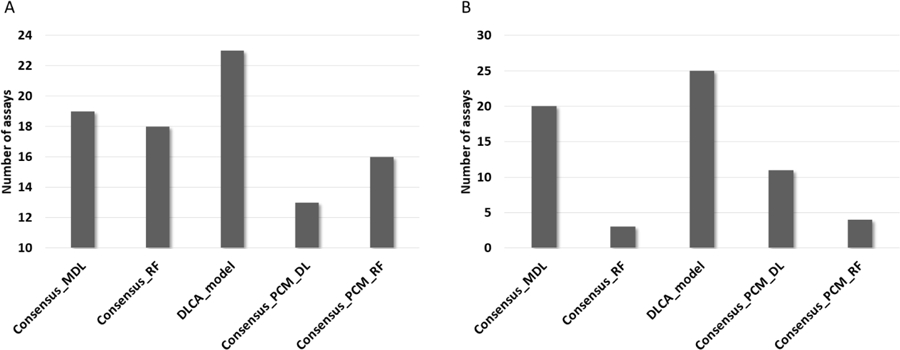
Assays applicability for 5 consensus models based on Tox21 data set.
(A) Number of assays predicted with AUC >= 0.85 calculated for external test set. (B) Number of assays predicted with BA >= 0.65 calculated for external test set.
According to the results in Figure 6, Consensus_MDL predicted about 50% of assays within the selected thresholds for the both AUC and BA values. The best results were found for DCLA model, which covered roughly 60% of assays within selected accuracy thresholds. Three remaining approaches showed inverse results for AUC and BA. Though, the PCM approaches covered 30–40% of assays. It is interesting to emphasize that only the DLCA method showed constantly good results across all metrics: RMSE, R2, AUC and BA, and highlights the benefit of finding the best consensus model during the back propagation.
Scaffold out validation strategy
In addition to the random splitting validation procedure we performed a scaffold out validation strategy to estimate the prediction power of models obtained with molecules of unseen scaffolds, which was suggested in the current studies.47 To do that we eliminated all compounds in ChEMBL and Tox21 test sets which share a common scaffold with compounds in the corresponding training sets. We then calculated the accuracy of prediction of consensus models using the constructed scaffold out test sets. The obtained results are presented in Table 3.
Table 3.
Accuracy of prediction for the developed 5 consensus models based on ChEMBL and Tox21 scaffold out test sets.
| Average RMSE value, ChEMBL data | Average AUC value, Tox21 data | |
|---|---|---|
| Consensus_MDL | 0.681 | 0.757 |
| Consensus_RF | 0.677 | 0.747 |
| DLCA_model | 0.665 | 0.774 |
| Consensus_PCM_DL | 0.673 | 0.758 |
| Consensus_PCM_RF | 0.713 | 0.754 |
Average root mean square error (RMSE) values calculated across all targets for scaffold out ChEMBL test set. Average AUC values calculated across all assays for scaffold out Tox21 test set. The best results are shown in bold.
As seen in Table 3, all methods performed worse compared to random splitting results. A decrease in prediction accuracy is expected with the scaffold out strategy as it undercuts the basic idea of QSAR and that similar compounds have the similar activity. The elimination of all compounds sharing a scaffolds forces significant extrapolation. However, the best results were still achieved by DCLA model on both ChEMBL and Tox21 scaffold out test sets. In fact, the overall rank order of models is still the same as in Table 1 for ChMEBL test set and almost the same as in Table 2 for Tox21 test set. The only differences were found that Consensus_PCM_DL showed the better results compared to Consensus_RF and Consensus_PCM_RF approaches. These might be explained by the fact that deep learning has better extrapolation ability compared to tree-based approaches.37
Influence of the applicability domain on prediction results
The estimation of a model’s applicability domain (AD) is a critical part of QSAR methodology. It was shown48,49 that utilization of AD can significantly improve the prediction results. In this study, we analyzed the influence of AD on the prediction results obtained from DLCA models. For assessment of applicability domain we used Tanimoto similarity based on Morgan fingerprints between test set compound and nearest neighbor in the training set. The corresponding calculations were performed separately for each particular target and assay. Thus, we calculated the similarity values for all compounds in the ChEMBL and Tox21 test sets and filtered out those compounds which were below the certain threshold. The prediction results were compared for both ChEMBL and Tox21 test sets considering the different cut-off values. Since AD limits the number of compounds for which model can be applied we also calculated coverage of prediction as percentage of compounds which fall in model’s AD. The distribution of R2 results for ChEMBL and BA for Tox21 test sets over AD cut-offs and corresponding coverage values are presented in Figure 7. The distribution of RMSE and AUC values were omitted in Figure 7 since they highly correlated with R2 and BA, respectively, for DLCA model.
Figure 7.
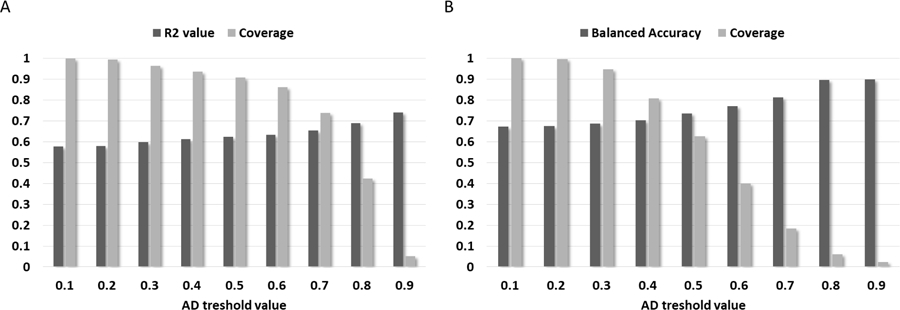
Distribution of the DLCA’s prediction results over AD cut-offs and coverage values.
(A) ChEMBL test set results. (B) Tox21 test set results.
The trend presented in Figure 7 shows a good correlation between model’s AD and prediction accuracy. Indeed, the higher AD threshold value the better accuracy of model’s prediction. However, the coverage of prediction is anti-correlated with AD and thus it is dramatically decreasing with increasing of AD values. The best prediction results were achieved with AD = 0.9, resulting in ChEMBL R2 = 0.741 and Tox21 BA = 0.898. Despite that, the coverage of prediction was small: ChEMBL coverage = 5% and Tox21 coverage = 2%. Results achieved with cut-off less than 0.3 are not significantly better than the original one. Considering both the accuracy of prediction and coverage we found that 0.5 cut-off provides the optimal ratio between them resulting in R2 = 0.623, coverage = 91% and BA = 0.735, coverage = 63% for ChEMBL and Tox21 test sets, respectively. Thus, compounds with similarity values falling in the region of 1 to 0.5 are considered to have the reasonable prediction results. Since there is a clear trend between accuracy of prediction and AD values, the utilized AD approach can be used for selection of compounds with certain confidence level of prediction.
Comparison of proposed consensus architecture with state of the art QSAR models
The developed deep learning consensus architecture is descriptor agnostic, and can be used to integrate of any type of descriptors and methods such as fingerprints, physical chemical properties, smiles50 and graph-convolutional based networks.51 In this study we incorporated only three types of fingerprints which were actively used over the decades by scientific community and showed benefits in computational drug discovery. It has previously been shown11 that models based on fingerprints/descriptors and neural nets can outperform the graph-convolution and smiles based models. However, as a proof of concept, we also compared the proposed approach with state of the art models implemented in MoleculeNet framework. We use the similar splitting of Tox21 challenge data set from MoleculeNet as was reported in the study of Wu et al.47 Since our approach is descriptor agnostic, we incorporated two extra models one by one to demonstrate this extensibility. We added the following models:
4 layers dense neural net based on 117 descriptors calculated using RDkit package.26 We named it “Tox21_DLCA_Desc”. All descriptors were normalized using Z-score transform.
SMILES based bidirectional LSTM model with Bahdanau attention mechanism.52 To convert smiles into numerical vector we used the tokenization approach implemented in Keras.33 We named the model as “Tox21_DLCA_Desc_LSTM”.
As was described previously, the 5-Fold CV procedure was used to established hyperparameters for the original DLCA model as well as for each model’s extension. First, we rebuilt the original DLCA model using only MoleculeNet’s Tox21 challenge training set, which consist of 12 end-points. The best accuracy was found with the following values: i) number of hidden layers {4}, ii) number of neurons for each layer {4000, 2000, 1500, 500}, iii) learning rate {0.001}, iv) activation function {ReLU}, v) batch size {32}, and vi) number of epochs {20}. We called this model “Tox21_DLCA”. Second, we added the descriptors model into Tox21_DLCA (and named as “Tox21_DLCA_Desc”). The best accuracy was found with the following values: i) number of hidden layers {4}, ii) number of neurons for each layer {1000, 700, 500, 300}, iii) learning rate {0.001}, iv) activation function {ReLU}, v) batch size {32}, and vi) number of epochs {10}. The selected parameters were used to construct Tox21_DLCA_Desc model using the entire Tox21 challenge training set from MoleculeNet. Third, we continued extension and added bidirectional LSTM model with following best parameters found during 5-Fold CV: i) number of hidden layers {4}, ii) types of hidden layers {Embedding, bidirectional LSTM , Bahdanau attention, Dense}, iii) number of neurons for each layer {100x128, 128, 128, 200}, iii) learning rate {0.002}, iv) activation function {ReLU}, v) batch size {32}, and vi) number of epochs {6}. The selected parameters were used to construct the final extension of DLCA model called “Tox21_DLCA_Desc_LSTM” using the same Tox21 challenge training set. Thus, totally we construct three DLCA models: i) the original one which is based on three RDkit fingerprints, ii) original model plus RDkit descriptors based model, iii) original model with RDkit descriptors based model and SMILES based bidirectional LSTM model with Bahdanau attention mechanism. We compared the developed three models with 9 different models described in the study of Wu et al on MoleculeNet’s Tox21 challenge test set. The comparison results are presented in Figure 8.
Figure 8.
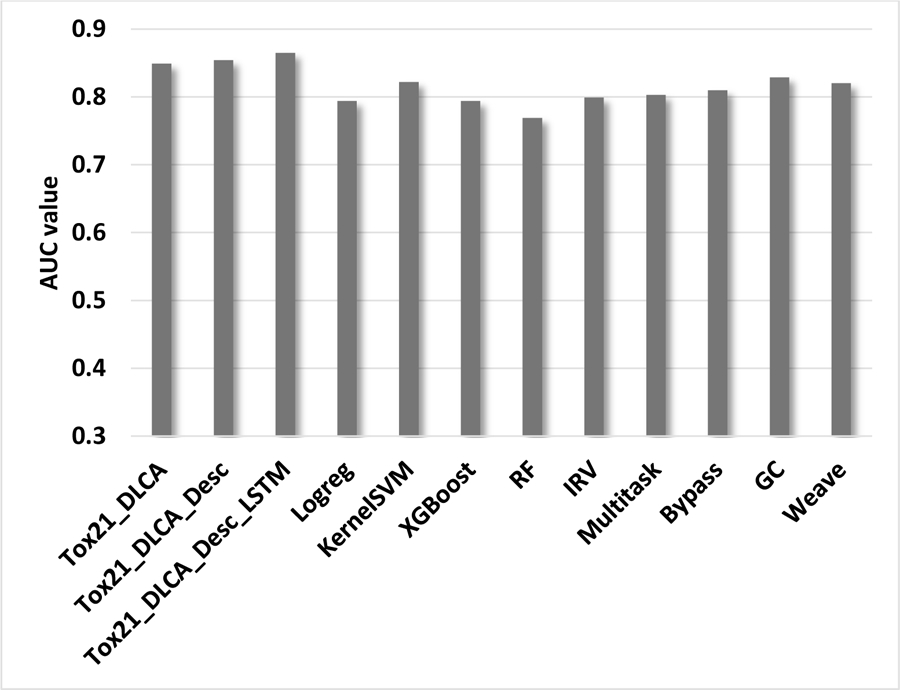
Comparison of three DLCA’s based models results with state of the arts approaches calculated as average AUC values for 12 end-point of Tox21 challenge data.
According to the average AUC values results in Figure 8, the original DLCA model as well as its extensions outperformed other models implemented in MoleculeNet for the 12 end-points of the Tox21 challenge data. The obtained results are expected since DLCA models incorporate more types of descriptors compared to MoleculeNet models. This study indicates the improvement of DLCA model performance by adding a new type of descriptor. Although thousands of descriptors exist and can be easily calculated, a comprehensive search of the best combination of descriptors types which can be incorporated into DLCA model is out of scope of this present study.
On-line service for prediction of biological activities profile for chemical compounds
DLCA models which were developed using ChEMBL and Tox21 data, are freely available on-line through a service named NCATS Predictor: https://predictor.ncats.io/. The web service we created provides both the predictive models and modeling sets, which were used to build the model. NCATS Predictor allows simultaneous prediction of different biological activities and physical-chemical properties of compounds. The service takes as input the different types of structure identifiers: Smiles, SDF files, Images of chemical structures. As output, the service provides predictions from the models calculated for each submitted compound. In addition, the service estimates and reports the applicability domain of each QSAR model. This calculation is performed for each compound with the result that each prediction is annotated with either “high”, “medium” or “ low” confidence, indicating whether one can be confident in the prediction or not. This annotation is based on Tanimoto similarity value calculated for nearest compound in the training set. Thus, a compound with similarity value to nearest neighbor falling in the region of 1 to 0.7 is considering to be predicted with a high confidence. If similarity value falls in the region of 0.7 to 0.5 then the compound is considered to be predicted with medium confidence. The obtained prediction results are considered to have a low confidence if similarity of compound is less than 0.5. The developed web service can be used by researchers for virtual screening of drug-like compounds with desirable biological profile as well as for structure optimization of the compound of interest. In addition to service, a self-contained instance of NCATS Predictor is available at: https://hub.docker.com/r/ncats/predx/.
CONCLUSIONS AND FUTURE DIRECTIONS
In this study, we have collected a variety of continuous and binary datasets from ChEMBL and Tox21 resources. We compared large-scale QSAR modeling approaches using two challenging datasets: i) more than quarter millions of compounds with quantitative data annotated across 1082 targets and ii) binary data obtained from 39 Tox21 assays. Proteochemometrics (PCM), Multitask Deep Learning (MDL), and conventional QSAR models were analyzed and compared for regression and classification tasks. We used three types of fingerprints: Morgan, Avalon, and AtomPair and two types of machine learning methods: deep learning and Random Forest. In addition to molecular fingerprints for PCM models we also used the target descriptors implemented in PROFEAT web service. Cross comparison of the conventional QSAR models revealed that each type of fingerprint performs differently depending on the machine learning approach, emphasizing the importance of utilizing of different descriptors types for successful modeling. It was also revealed that concatenation of descriptors did not improve the prediction results compared to separate models. We explored consensus modeling which integrates outputs from models based on different descriptors. A consensus modeling strategy was applied for Proteochemometrics (PCM), Multitask Deep Learning (MDL), and conventional QSAR methods. Consensus_PCM, consensus_MDL and consensus_RF showed the good accuracy of prediction on ChEMBL data. However, from these three approaches only consensus_MDL performed reasonably well on Tox21 data. We have proposed and developed a new deep learning architecture which allows not only perform transfer knowledge across different target/assays, but also capable to find the best way to incorporate contributions from models based on different descriptors. We call this approach DLCA (deep learning consensus architecture). The DLCA method demonstrated the best performance for both regression and classification tasks compared to other consensus approaches. It is interesting to emphasize that any type of descriptors can be incorporated into DLCA model. In this study, we utilized fingerprints, but since the architecture has modular basis it can be easy extended to any other descriptors such as physical-chemical properties, fragmental descriptors, etc. Also, the modular nature of DLCA system allows to incorporate even different deep learning approaches like graph-convolution models51 or recurrent neural nets with SMILES as descriptors.53 To demonstrate that concept we extend the DLCA architecture by adding RDkit descriptors and the SMILES based bidirectional LSTM model and compared this extension with state of the art machine learning/deep learning approaches implemented in MoleculeNet using 12 end-point from Tox21 challenge data set. The original DLCA model as well as its extensions outperformed both other models. In addition to performance comparison, we analyzed the influence of applicability domain (AD) of DLCA model and found the good correlation between accuracy of model and calculated AD values. The utilized AD approach can be used for flexible selection of compounds with certain confidence level of prediction. We have disseminated the DLCA models together with modeling sets through publicly available web service and also as self-contained instance through the docker image.
Supplementary Material
ACKNOWLEDGMENT
This project was funded in part with Intramural Research Program, National Center for Advancing Translational Sciences, National Institutes of Health (1ZIATR000058–03).
ABBREVIATIONS
- QSAR
quantitative structure-activity relationships
- MDL
multitask deep learning
- PCM
protochemometrics
- DCLA
deep learning consensus architecture
- DL
deep learning
- RF
random forest
- AD
applicability domain
- RMSE
root mean square error
- BA
balanced accuracy
- AUC
area under the receiver operating characteristic curve
Footnotes
ASSOCIATED CONTENT
Supporting Information
The performance of developed models across ChEMBL targets and Tox21 assays are available in the Supplementary Material.
The authors declare no competing financial interest.
REFERENCES
- (1).Oprea TI; Tropsha A Target, Chemical and Bioactivity Databases – Integration Is Key. Drug Discov. Today Technol 2006, 3 (4), 357–365. 10.1016/j.ddtec.2006.12.003. [DOI] [Google Scholar]
- (2).Brooksbank C; Bergman MT; Apweiler R; Birney E; Thornton J The European Bioinformatics Institute’s Data Resources 2014. Nucleic Acids Res 2014, 42 (Database issue), D18–D25. 10.1093/nar/gkt1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gaulton A; Bellis LJ; Bento AP; Chambers J; Davies M; Hersey A; Light Y; McGlinchey S; Michalovich D; Al-Lazikani B; Overington JP ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res 2011, 40 (D1), D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bolton EE; Wang Y; Thiessen PA; Bryant SH Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annual Reports in Computational Chemistry; Wheeler Ralph A. and Spellmeyer David C., Ed.; Elsevier, 2008; Vol. Volume 4, pp 217–241. [Google Scholar]
- (5).Koutsoukas A; Lowe R; KalantarMotamedi Y; Mussa HY; Klaffke W; Mitchell JBO; Glen RC; Bender A In Silico Target Predictions: Defining a Benchmarking Data Set and Comparison of Performance of the Multiclass Naïve Bayes and Parzen-Rosenblatt Window. J. Chem. Inf. Model 2013, 53 (8), 1957–1966. 10.1021/ci300435j. [DOI] [PubMed] [Google Scholar]
- (6).Pogodin PV; Lagunin AA; Filimonov DA; Poroikov VV PASS Targets: Ligand-Based Multi-Target Computational System Based on a Public Data and Naïve Bayes Approach. SAR QSAR Environ. Res 2015, 26 (10), 783–793. 10.1080/1062936X.2015.1078407. [DOI] [PubMed] [Google Scholar]
- (7).Clark AM; Ekins S Open Source Bayesian Models. 2. Mining a “Big Dataset” To Create and Validate Models with ChEMBL. J. Chem. Inf. Model 2015, 55 (6), 1246–1260. 10.1021/acs.jcim.5b00144. [DOI] [PubMed] [Google Scholar]
- (8).Varnek A; Gaudin C; Marcou G; Baskin I; Pandey AK; Tetko IV Inductive Transfer of Knowledge: Application of Multi-Task Learning and Feature Net Approaches to Model Tissue-Air Partition Coefficients. J. Chem. Inf. Model 2009, 49 (1), 133–144. 10.1021/ci8002914. [DOI] [PubMed] [Google Scholar]
- (9).Ramsundar B; Kearnes S; Riley P; Webster D; Konerding D; Pande V Massively Multitask Networks for Drug Discovery. ArXiv150202072 Cs Stat 2015. [Google Scholar]
- (10).Kearnes S; Goldman B; Pande V Modeling Industrial ADMET Data with Multitask Networks. ArXiv160608793 Stat 2016. [Google Scholar]
- (11).Mayr A; Klambauer G; Unterthiner T; Steijaert M; Wegner JK; Ceulemans H; Clevert D-A; Hochreiter S Large-Scale Comparison of Machine Learning Methods for Drug Target Prediction on ChEMBL. Chem. Sci 2018, 9 (24), 5441–5451. 10.1039/C8SC00148K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ramsundar B; Liu B; Wu Z; Verras A; Tudor M; Sheridan RP; Pande V Is Multitask Deep Learning Practical for Pharma? J. Chem. Inf. Model 2017, 57 (8), 2068–2076. 10.1021/acs.jcim.7b00146. [DOI] [PubMed] [Google Scholar]
- (13).Xu Y; Ma J; Liaw A; Sheridan RP; Svetnik V Demystifying Multitask Deep Neural Networks for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model 2017, 57 (10), 2490–2504. 10.1021/acs.jcim.7b00087. [DOI] [PubMed] [Google Scholar]
- (14).Cortés-Ciriano I; Ain QU; Subramanian V; Lenselink EB; Méndez-Lucio O; IJzerman AP; Wohlfahrt G; Prusis P; Malliavin TE; Westen G. J. P. van; Bender A Polypharmacology Modelling Using Proteochemometrics (PCM): Recent Methodological Developments, Applications to Target Families, and Future Prospects. MedChemComm 2015, 6 (1), 24–50. 10.1039/C4MD00216D. [DOI] [Google Scholar]
- (15).Ain QU; Méndez-Lucio O; Ciriano IC; Malliavin T; Westen G. J. P. van; Bender A Modelling Ligand Selectivity of Serine Proteases Using Integrative Proteochemometric Approaches Improves Model Performance and Allows the Multi-Target Dependent Interpretation of Features. Integr. Biol 2014, 6 (11), 1023–1033. 10.1039/C4IB00175C. [DOI] [PubMed] [Google Scholar]
- (16).Cortés-Ciriano I; van Westen GJP; Bouvier G; Nilges M; Overington JP; Bender A; Malliavin TE Improved Large-Scale Prediction of Growth Inhibition Patterns Using the NCI60 Cancer Cell Line Panel. Bioinformatics 2016, 32 (1), 85–95. 10.1093/bioinformatics/btv529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cheng F; Zhou Y; Li J; Li W; Liu G; Tang Y Prediction of Chemical–protein Interactions: Multitarget-QSAR versus Computational Chemogenomic Methods. Mol. Biosyst 2012, 8 (9), 2373–2384. 10.1039/C2MB25110H. [DOI] [PubMed] [Google Scholar]
- (18).Lenselink EB; ten Dijke N; Bongers B; Papadatos G; van Vlijmen HWT; Kowalczyk W; IJzerman AP; van Westen GJP Beyond the Hype: Deep Neural Networks Outperform Established Methods Using a ChEMBL Bioactivity Benchmark Set. J. Cheminformatics 2017, 9, 45. 10.1186/s13321-017-0232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zakharov AV; Peach ML; Sitzmann M; Nicklaus MC QSAR Modeling of Imbalanced High-Throughput Screening Data in PubChem. J. Chem. Inf. Model 2014, 54 (3), 705–712. 10.1021/ci400737s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhu H; Tropsha A; Fourches D; Varnek A; Papa E; Gramatica P; Öberg T; Dao P; Cherkasov A; Tetko IV Combinatorial QSAR Modeling of Chemical Toxicants Tested against Tetrahymena Pyriformis. J. Chem. Inf. Model 2008, 48 (4), 766–784. 10.1021/ci700443v. [DOI] [PubMed] [Google Scholar]
- (21).Zakharov AV; Varlamova EV; Lagunin AA; Dmitriev AV; Muratov EN; Fourches D; Kuz’min VE; Poroikov VV; Tropsha A; Nicklaus MC QSAR Modeling and Prediction of Drug–Drug Interactions. Mol. Pharm 2016, 13 (2), 545–556. 10.1021/acs.molpharmaceut.5b00762. [DOI] [PubMed] [Google Scholar]
- (22).Xu Y; Pei J; Lai L Deep Learning Based Regression and Multiclass Models for Acute Oral Toxicity Prediction with Automatic Chemical Feature Extraction. J. Chem. Inf. Model 2017, 57 (11), 2672–2685. 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- (23).Martin EJ; Polyakov VR; Tian L; Perez RC Profile-QSAR 2.0: Kinase Virtual Screening Accuracy Comparable to Four-Concentration IC50s for Realistically Novel Compounds. J. Chem. Inf. Model 2017, 57 (8), 2077–2088. 10.1021/acs.jcim.7b00166. [DOI] [PubMed] [Google Scholar]
- (24).Toxicology in the 21st Century (Tox21) https://ncats.nih.gov/tox21 (accessed Feb 25, 2019).
- (25).Tox21 Data Browser https://tripod.nih.gov/tox21 (accessed Feb 25, 2019).
- (26).RDKit http://www.rdkit.org/ (accessed Apr 27, 2016).
- (27).Rogers D; Hahn M Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- (28).Gedeck P; Rohde B; Bartels C QSAR − How Good Is It in Practice? Comparison of Descriptor Sets on an Unbiased Cross Section of Corporate Data Sets. J. Chem. Inf. Model 2006, 46 (5), 1924–1936. 10.1021/ci050413p. [DOI] [PubMed] [Google Scholar]
- (29).Carhart RE; Smith DH; Venkataraghavan R Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J. Chem. Inf. Comput. Sci 1985, 25 (2), 64–73. 10.1021/ci00046a002. [DOI] [Google Scholar]
- (30).Zhang Q; Muegge I Scaffold Hopping through Virtual Screening Using 2D and 3D Similarity Descriptors: Ranking, Voting, and Consensus Scoring. J. Med. Chem 2006, 49 (5), 1536–1548. 10.1021/jm050468i. [DOI] [PubMed] [Google Scholar]
- (31).Zhang P; Tao L; Zeng X; Qin C; Chen S; Zhu F; Li Z; Jiang Y; Chen W; Chen Y-Z A Protein Network Descriptor Server and Its Use in Studying Protein, Disease, Metabolic and Drug Targeted Networks. Brief. Bioinform 2017, 18 (6), 1057–1070. 10.1093/bib/bbw071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).scikit-learn: machine learning in Python — scikit-learn 0.20.2 documentation https://scikit-learn.org/stable/ (accessed Jan 8, 2019).
- (33).Keras Documentation https://keras.io/ (accessed Mar 3, 2017).
- (34).TensorFlow https://www.tensorflow.org/ (accessed Feb 14, 2018).
- (35).Kingma DP; Ba J Adam: A Method for Stochastic Optimization. ArXiv14126980 Cs 2014. [Google Scholar]
- (36).Popova M; Isayev O; Tropsha A Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv 2018, 4 (7), eaap7885. 10.1126/sciadv.aap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ma J; Sheridan RP; Liaw A; Dahl GE; Svetnik V Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model 2015, 55 (2), 263–274. 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- (38).Dahl GE; Jaitly N; Salakhutdinov R Multi-Task Neural Networks for QSAR Predictions. ArXiv14061231 Cs Stat 2014. [Google Scholar]
- (39).Klambauer G; Unterthiner T; Mayr A; Hochreiter S Self-Normalizing Neural Networks 2017. [Google Scholar]
- (40).Fawcett T An Introduction to ROC Analysis. Pattern Recognit. Lett 2006, 27 (8), 861–874. 10.1016/j.patrec.2005.10.010. [DOI] [Google Scholar]
- (41).Huang R; Xia M; Nguyen D-T; Zhao T; Sakamuru S; Zhao J; Shahane SA; Rossoshek A; Simeonov A Tox21Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways as Mediated by Exposure to Environmental Chemicals and Drugs. Front. Environ. Sci 2016, 3. 10.3389/fenvs.2015.00085. [DOI] [Google Scholar]
- (42).Mayr A; Klambauer G; Unterthiner T; Hochreiter S DeepTox: Toxicity Prediction Using Deep Learning. Front. Environ. Sci 2016, 3. 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- (43).Tox21 Data Challenge 2014https://tripod.nih.gov/tox21/challenge/ (accessed Nov 28, 2017).
- (44).Golbraikh A; Tropsha A Beware of Q2! J. Mol. Graph. Model 2002, 20 (4), 269–276. 10.1016/S1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- (45).Mansouri K; Abdelaziz A; Rybacka A; Roncaglioni A; Tropsha A; Varnek A; Zakharov A; Worth A; Richard AM; Grulke CM; Trisciuzzi D; Fourches D; Horvath D; Benfenati E; Muratov E; Wedebye EB; Grisoni F; Mangiatordi GF; Incisivo GM; Hong H; Ng HW; Tetko IV; Balabin I; Kancherla J; Shen J; Burton J; Nicklaus M; Cassotti M; Nikolov NG; Nicolotti O; Andersson PL; Zang Q; Politi R; Beger RD; Todeschini R; Huang R; Farag S; Rosenberg SA; Slavov S; Hu X; Judson RS CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect 2016, 124 (7), 1023–1033. 10.1289/ehp.1510267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lagunin A; Zakharov A; Filimonov D; Poroikov V QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Mol. Inform 2011, 30 (2–3), 241–250. 10.1002/minf.201000151. [DOI] [PubMed] [Google Scholar]
- (47).Wu Z; Ramsundar B; Feinberg EN; Gomes J; Geniesse C; Pappu AS; Leswing K; Pande V MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci 2018, 9 (2), 513–530. 10.1039/C7SC02664A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tetko IV; Sushko I; Pandey AK; Zhu H; Tropsha A; Papa E; Öberg T; Todeschini R; Fourches D; Varnek A Critical Assessment of QSAR Models of Environmental Toxicity against Tetrahymena Pyriformis: Focusing on Applicability Domain and Overfitting by Variable Selection. J. Chem. Inf. Model 2008, 48 (9), 1733–1746. 10.1021/ci800151m. [DOI] [PubMed] [Google Scholar]
- (49).Sushko I; Novotarskyi S; Körner R; Pandey AK; Cherkasov A; Li J; Gramatica P; Hansen K; Schroeter T; Müller K-R; Xi L; Liu H; Yao X; Öberg T; Hormozdiari F; Dao P; Sahinalp C; Todeschini R; Polishchuk P; Artemenko A; Kuz’min V; Martin TM; Young DM; Fourches D; Muratov E; Tropsha A; Baskin I; Horvath D; Marcou G; Muller C; Varnek A; Prokopenko VV; Tetko IV Applicability Domains for Classification Problems: Benchmarking of Distance to Models for Ames Mutagenicity Set. J. Chem. Inf. Model 2010, 50 (12), 2094–2111. 10.1021/ci100253r. [DOI] [PubMed] [Google Scholar]
- (50).Jastrzębski S; Leśniak D; Czarnecki WM Learning to SMILE(S). ArXiv160206289 Cs 2016. [Google Scholar]
- (51).Kearnes S; McCloskey K; Berndl M; Pande V; Riley P Molecular Graph Convolutions: Moving beyond Fingerprints. J. Comput. Aided Mol. Des 2016, 30 (8), 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bahdanau D; Cho K; Bengio Y Neural Machine Translation by Jointly Learning to Align and Translate 2014. [Google Scholar]
- (53).Bjerrum EJ; Sattarov B Improving Chemical Autoencoder Latent Space and Molecular De Novo Generation Diversity with Heteroencoders. Biomolecules 2018, 8 (4). 10.3390/biom8040131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.