Published on Web 03/15/2010
Ni(III)/(IV) Bis(dicarbollide) as a Fast, Noncorrosive Redox Shuttle for
Dye-Sensitized Solar Cells
Tina C. Li,†,‡ Alexander M. Spokoyny,†,§ Chunxing She,†,‡ Omar K. Farha,†,§ Chad A. Mirkin,*,†,§
Tobin J. Marks,*,†,‡ and Joseph T. Hupp*,†,‡,§
Department of Chemistry, Argonne-Northwestern Solar Energy Research Center (ANSER), and International
Institute for Nanotechnology, Northwestern UniVersity, 2145 Sheridan Road, EVanston, Illinois 60208
Received January 15, 2010; E-mail: j-hupp@northwestern.edu; t-marks@northwestern.edu; chadnano@northwestern.edu
The favorable energetics of dye-sensitized solar cell (DSC)
constituents have advanced photoelectrochemical power conversion
efficiencies to an impressive ∼11%.1 However, the performance
of DSCs based on the I-/I3- redox couple has essentially plateaued,
and additional component modifications have not advanced efficiency.2 Indeed, the limitations placed on DSC dyes, semiconductors, and counter electrodes by I-/I3- compatibility have presented
a major challenge. DSC efficiency rests on a delicate balance of
charge transport and recombination processes, with attempts to find
redox shuttle alternatives to the I-/I3- meeting limited success. To
date, only a handful of cationic redox couples (e.g., Co(II/III)3 and
Cu(I/II)4 complexes) and p-type semiconductors5 have functioned
as effective redox mediators. While rapidly exchanging, outersphere redox couples such as ferrocene/ferrocenium (Fc/Fc+) appear,
a priori, promising for dyes with low overpotentials, rapid interception of injected electrons by Fc+ leads to inadequate electron
diffusion lengths and charge lifetimes.6
In searching for chemically robust and DSC-compatible redox
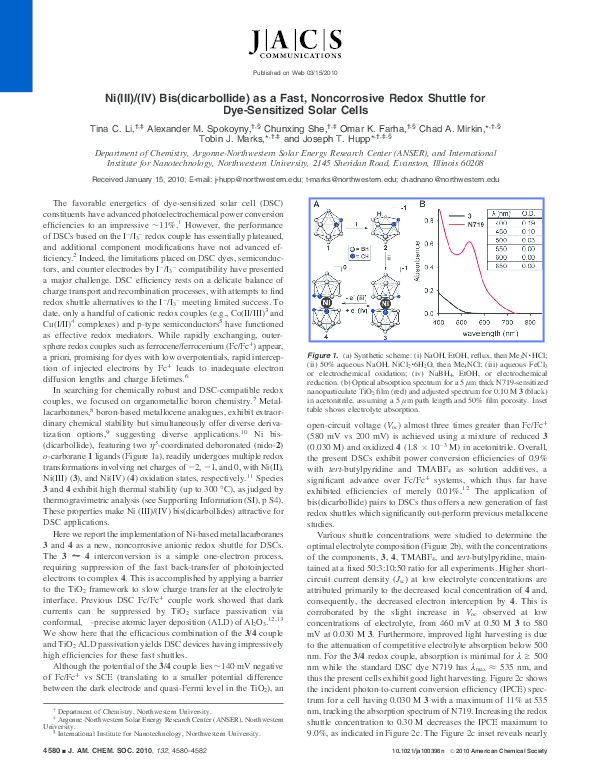
couples, we focused on organometallic boron chemistry.7 Metallacarboranes,8 boron-based metallocene analogues, exhibit extraordinary chemical stability but simultaneously offer diverse derivatization options,9 suggesting diverse applications.10 Ni bis(dicarbollide), featuring two η5-coordinated deboronated (nido-2)
o-carborane 1 ligands (Figure 1a), readily undergoes multiple redox
transformations involving net charges of -2, -1, and 0, with Ni(II),
Ni(III) (3), and Ni(IV) (4) oxidation states, respectively.11 Species
3 and 4 exhibit high thermal stability (up to 300 °C), as judged by
thermogravimetric analysis (see Supporting Information (SI), p S4).
These properties make Ni (III)/(IV) bis(dicarbollides) attractive for
DSC applications.
Here we report the implementation of Ni-based metallacarboranes
3 and 4 as a new, noncorrosive anionic redox shuttle for DSCs.
The 3 H 4 interconversion is a simple one-electron process,
requiring suppression of the fast back-transfer of photoinjected
electrons to complex 4. This is accomplished by applying a barrier
to the TiO2 framework to slow charge transfer at the electrolyte
interface. Previous DSC Fc/Fc+ couple work showed that dark
currents can be suppressed by TiO2 surface passivation via
conformal, Å-precise atomic layer deposition (ALD) of Al2O3.12,13
We show here that the efficacious combination of the 3/4 couple
and TiO2 ALD passivation yields DSC devices having impressively
high efficiencies for these fast shuttles.
Although the potential of the 3/4 couple lies ∼140 mV negative
of Fc/Fc+ vs SCE (translating to a smaller potential difference
between the dark electrode and quasi-Fermi level in the TiO2), an
†
Department of Chemistry, Northwestern University.
Argonne-Northwestern Solar Energy Research Center (ANSER), Northwestern
University.
§
International Institute for Nanotechnology, Northwestern University.
‡
4580
9
J. AM. CHEM. SOC. 2010, 132, 4580–4582
Figure 1. (a) Synthetic scheme: (i) NaOH, EtOH, reflux, then Me3N · HCl;
(ii) 50% aqueous NaOH, NiCl2 · 6H2O, then Me4NCl; (iii) aqueous FeCl3
or electrochemical oxidation; (iv) NaBH4, EtOH, or electrochemical
reduction. (b) Optical absorption spectrum for a 5 µm thick N719-sensitized
nanoparticulate TiO2 film (red) and adjusted spectrum for 0.10 M 3 (black)
in acetonitrile, assuming a 5 µm path length and 50% film porosity. Inset
table shows electrolyte absorption.
open-circuit voltage (Voc) almost three times greater than Fc/Fc+
(580 mV vs 200 mV) is achieved using a mixture of reduced 3
(0.030 M) and oxidized 4 (1.8 × 10-3 M) in acetonitrile. Overall,
the present DSCs exhibit power conversion efficiencies of 0.9%
with tert-butylpyridine and TMABF4 as solution additives, a
significant advance over Fc/Fc+ systems, which thus far have
exhibited efficiencies of merely 0.01%.12 The application of
bis(dicarbollide) pairs to DSCs thus offers a new generation of fast
redox shuttles which significantly out-perform previous metallocene
studies.
Various shuttle concentrations were studied to determine the
optimal electrolyte composition (Figure 2b), with the concentrations
of the components, 3, 4, TMABF4, and tert-butylpyridine, maintained at a fixed 50:3:10:50 ratio for all experiments. Higher shortcircuit current density (Jsc) at low electrolyte concentrations are
attributed primarily to the decreased local concentration of 4 and,
consequently, the decreased electron interception by 4. This is
corroborated by the slight increase in Voc observed at low
concentrations of electrolyte, from 460 mV at 0.50 M 3 to 580
mV at 0.030 M 3. Furthermore, improved light harvesting is due
to the attenuation of competitive electrolyte absorption below 500
nm. For the 3/4 redox couple, absorption is minimal for λ g 500
nm while the standard DSC dye N719 has λmax ≈ 535 nm, and
thus the present cells exhibit good light harvesting. Figure 2c shows
the incident photon-to-current conversion efficiency (IPCE) spectrum for a cell having 0.030 M 3 with a maximum of 11% at 535
nm, tracking the absorption spectrum of N719. Increasing the redox
shuttle concentration to 0.30 M decreases the IPCE maximum to
9.0%, as indicated in Figure 2c. The Figure 2c inset reveals nearly
10.1021/ja100396n 2010 American Chemical Society
�COMMUNICATIONS
Figure 2. (a) J-V characteristics of DSCs containing 3 (0.030 M), 4 (1.8 × 10-3 M), TMABF4 (6.0 × 10-3 M), and tert-butylpyridine (0.036 M) with 0-2
cycles of ALD Al2O3 coverage of the TiO2; (b) J-V characteristics with decreasing electrolyte concentration; (c) IPCE comparison of electrolyte containing
0.30 M (blue dotted) and 0.030 M (black solid) TMA-3 solutions (inset shows normalized IPCE spectra).
identical photoaction spectra by scaling the IPCE spectrum of the
0.30 M cell by 1.26×, indicating minor photon loss due to
electrolyte absorption. This 21% IPCE loss is attributed to the 10fold increase in redox shuttle concentration and is confirmed by a
parallel decrease in Jsc, from 2.4 mA/cm2 at 0.030 M to 1.9 mA/
cm2 at 0.30 M of 3.
Thin insulating layers of large band gap MgO,14 ZrO2,15
SrTiO3,16 and Al2O317 have been previously deposited on DSC TiO2
photoelectrodes to tune interfacial charge dynamics. Electron
interception by the electrolyte is considered to be the predominant
power conversion pathway, assuming fast dye regeneration. With
a physical barrier at the TiO2/dye interface, charge leakage resulting
from direct semiconductor-electrolyte contact is reduced, thus
dramatically enhancing overall DSC performance. Using ALD,
conformal Al2O3 growth is effectively achieved (∼1.1 Å per ALD
cycle) on the TiO2 nanoparticle framework. Increased Jsc is believed
to largely reflect passivation of surface states causing localization
of electron density on the TiO2 surface, since 1 Al2O3 ALD cycle
deposits less than a monolayer.12 Eliminating these defect sites
impedes electron capture by the neighboring redox couple, thus
reducing dark current. For each order of magnitude that charge
interception is reduced, there is a γ · 59 mV gain in photovoltage,
where the diode quality factor γ is usually 1.0-1.5, assuming
electron transfer from the conduction band.18 This effect is
confirmed by the increase in Voc from 580 mV to 640 mV with 1
cycle of Al2O3 passivation, yielding Jsc ) 3.76 mA/cm2 and an
overall power conversion efficiency of 1.5%. Further Al2O3
deposition hinders current collection, reducing Jsc by ∼50% for 2
Al2O3 cycles. This drop in current may indicate the suppression of
electron injection, resulting from the insulating Al2O3 barrier. With
2 ALD cycles, the TiO2 surface is completely coated with Al2O3.
Thus, beyond 1 deposition cycle, charge interception is no longer
reduced primarily by trap state passivation but by increased distance
between photoinjected electrons and 4. Retardation of electron
interception is apparent in the successive photovoltage increase with
increasing Al2O3 coverage, but optimal performance is achieved at
only 1 ALD cycle due to its relatively high Jsc.
The open-circuit voltage decay technique was next used to
probe the kinetics of interception by the electrolyte.19 Comparisons of the 3/4 redox couple with I-/I3- and Fc/Fc+ are shown
in Figure 3a with potentials adjusted to the Fc/Fc+ solution
potential for reference.12 From the charge lifetimes vs potential
plots, interception appears to be ∼103× slower than for Fc/Fc+,
but almost 100× faster than I-/I3-. Slower 3/4 shuttle kinetics
are expected since the Ni (IV) f Ni(III) reduction requires
dicarbollide rotation from a cis f trans conformation (see Figure
Figure 3. (a) Charge lifetime plots from Voc decay for different redox
shuttles with 1 ALD cycle of Al2O3 on the TiO2 nanoparticle framework.
(b) Ground-state recovery kinetics of N719 adsorbed on TiO2 films probed
at 500 nm after excitation at 532 nm. Films were soaked in 0.03 M of 3
and I- solutions.
1a). This in turn creates a larger activation barrier for the
reduction of 4 than for Fc+, translating to a slower interception
rate. However, compared to I-/I3-, the 3/4 kinetics are fast,
involving facile 3 f 4 one-electron transfer, thereby allowing
efficient dye regeneration, and opening the possibility of
incorporating DSC dyes with less positive ground state potentials
than widely used “N719”. Nevertheless, to make full use of the
new shuttle, it will be necessary to turn to photoelectrode
materials and architectures displaying faster electron transport
and collection;20 the observed modest IPCE values indicate, with
nanoparticulate TiO2 as the photoelectrode material, significant
deleterious competition from electron interception by 4.
Transient absorption spectroscopy confirms that the remarkable
performance of 0.030 M 3 results from fast dye regeneration. Thus,
Figure 3b compares N719 ground-state recovery kinetics, probed
at 500 nm, for 0.030 M 3, 0.030 M 1-butyl-3-methylimidazolium
iodide, and pure solvent. Dye regeneration in the absence of
electrolyte (recombination) occurs in tens of microseconds and
J. AM. CHEM. SOC.
9
VOL. 132, NO. 13, 2010
4581
�COMMUNICATIONS
slower, agreeing well with literature data.21 Dye regeneration by 3
occurs in <2 µs (t1/2), slightly faster than that by I-. Remarkably,
regeneration by 3 is 10-100× faster than recombination, thus
affording a regeneration yield of g90%.
In addition to efficient dye regeneration kinetics, the 3/4 shuttle
has a fast rate of electron exchange at the counter electrode,
minimizing voltage losses.22 Platinized counter electrodes are
typically used in DSCs since Pt efficiently catalyzes the I3- f Ireduction.23 However, Au and carbon have also been used as
alternative cathode materials for cobalt-based shuttles.4 Unlike the
I-/I3- redox mediator, the 3/4 shuttle is stable with respect to other
conductive metals, and in fact, replacing Pt with Ag or Au at the
counter electrodes gives similar or enhanced photovoltaic characteristics. Thus, a Au counter electrode gives the highest Jsc and fill
factor in this study (see SI, p S6), most likely due to the greater
electrode reflectivity, increasing the optical path through the cell.
The performance with Ag was also explored and shown to follow
similar trends with ALD modification. Other cost-efficient materials
are being explored.
In summary, the 3/4 shuttle is shown to be a promising,
noncorrosive DSC shuttle with good solubility and redox properties.
In contrast to Co and Cu based shuttles, 4 is neutrally charged and
therefore does not appreciably adsorb on the TiO2 photoanode.
Additionally, the 3/4 shuttle exhibits slower kinetics than Fc/Fc+
and therefore a slower rate of electron interception by the electrolyte.
Marked suppression of dark currents is achieved, with the kinetically
slower 3/4 reaching photovoltages of 640 mV after Al2O3 deposition, significantly higher than the case for the Fc/Fc+ couple, and
enhancing current densities by 2-3× with only 1 Al2O3 ALD cycle.
Furthermore, Au is shown to be an attractive alternative counter
electrode in these cells, providing superior current densities and
fill factors. The remarkable inertness of 3/4 toward Ag metal may
be beneficial for large-scale DSC fabrication, if Ag contacts are
used. This novel fast, one-electron transfer system opens the
possibility for new dyes and frameworks to be incorporated into
DSCs, with dicarbollide ligands readily tunable to alter the shuttle
charge transfer characteristics.
Acknowledgment. J.T.H. and T.J.M. gratefully acknowledge
the support of BP Solar. J.T.H. also thanks the U.S. DOE Office
of Science (Grant No. DE-FG02-87ER13808) and NU-NSEC for
funding. T.J.M. acknowledges the support of the Energy Frontier
Research center (DE-SC0001059) at the ANSER Center of
Northwestern University. C.A.M. acknowledges the DOE Office
of Basic Energy Sciences (Award No. DE-SC0000989) for support
via the NU Nonequilibrium Energy Research Center. He is also
grateful for support from the Army Research Office.
Supporting Information Available: Experimental data, solar cell
fabrication and characterizations, CV, TGA, current transients between
1/10 to 1 sun illuminations, and J-V characteristics for varying
electrolyte concentrations, alternative counter electrodes and with ALD
Al2O3 modification. This material is available free of charge via the
Internet at http://pubs.acs.org.
References
(1) (a) Grätzel, M. Inorg. Chem. 2005, 44, 6841. (b) Chiba, Y.; Islam, A.;
Watanabe, Y.; Komiya, R.; Koide, N.; Han, L. Y. Jpn. J. Appl. Phys. Pt.
2 2006, 45, L638–L640. (c) Boschloo, G.; Hagfeldt, A. Acc. Chem. Res.
2009, 42, 1819–1826.
(2) (a) Hamann, T. W.; Jensen, R. A.; Martinson, A. B. F.; Ryswyk, H. V.;
Hupp, J. T. Energy EnViron. Sci. 2008, 1, 66–78. (b) Martinson, A. B. F.;
4582
J. AM. CHEM. SOC.
9
VOL. 132, NO. 13, 2010
Hamann, T. W.; Pellin, M. J.; Hupp, J. T. Chem.sEur. J. 2008, 14, 4458–
4467.
(3) (a) Cameron, P. J.; Peter, L. M.; Zakeeruddin, S. M.; Grätzel, M. Coord.
Chem. ReV. 2004, 248, 1447–1453. (b) Cazzanti, S.; Caramori, S.; Argazzi,
R.; Elliot, C. M.; Bignozzi, C. A. J. Am. Chem. Soc. 2006, 128, 9996–
9997. (c) Sapp, S. A.; Elliott, C. M.; Contado, C.; Caramori, S.; Bignozzi,
C. A. J. Am. Chem. Soc. 2002, 124, 11215–11222.
(4) Hattori, S.; Wada, Y.; Yanagida, S.; Fukuzumi, S. J. Am. Chem. Soc. 2005,
127, 9648–9654.
(5) (a) Yum, J.-H.; Chen, P.; Grätzel, M.; Nazeeruddin, M. K. ChemSusChem
2008, 1, 699–707. (b) Li, B.; Wang, L.; Kang, B.; Wang, P.; Qiu, Y. Sol.
Energy Mater. Sol. Cells 2006, 90, 549–573. (c) Bach, U.; Lupo, D.; Comte,
P.; Moser, J. E.; Weissörtel, F.; Salbeck, J.; Spreitzer, H.; Grätzel, M. Nature
1998, 395, 583–585. (d) Yanagida, S.; Yu, Y.; Manseki, K. Acc. Chem.
Res. 2009, 42, 1827–1838.
(6) Gregg, B. A.; Pichot, F.; Ferrere, S.; Fields, C. L. J. Phys. Chem. B 2001,
105, 1422–1429.
(7) Siebert, W. AdVances in Boron Chemistry; UK, 1997.
(8) Grimes, R. N. Carboranes; Academic Press: New York, 1970.
(9) (a) Warren, L. F.; Hawthorne, M. F. J. Am. Chem. Soc. 1970, 92, 1157–
1173. (b) Farha, O. K.; Spokoyny, A. M.; Mulfort, K. L.; Hawthorne, M. F.;
Mirkin, C. A.; Hupp, J. T. J. Am. Chem. Soc. 2007, 129, 12680–12681. (c)
Wade, K. Nat. Chem. 2009, 1, 92. (d) Spokoyny, A. M.; Reuter, M. G.;
Stern, C. L.; Ratner, M. A.; Seideman, T.; Mirkin, C. A. J. Am. Chem.
Soc. 2009, 131, 9482–9483. (e) Jude, H.; Disteldorg, H.; Fischer, S.; Wedge,
T.; Hawkridge, A. M.; Arif, A. M.; Hawthorne, M. F.; Muddiman, D. C.;
Stang, P. J. J. Am. Chem. Soc. 2005, 127, 12131–12139. (f) Dash, B. P.;
Satapathy, R.; Maguire, J. A.; Hosmane, N. S. Org. Lett. 2008, 10, 22247–
2250. (g) Mueller, J.; Base, K.; Michl, J. J. Am. Chem. Soc. 1992, 114,
9721–9722.
(10) (a) Hawthorne, M. F.; Maderna, A. Chem. ReV. 1999, 99, 3421–3434. (b)
Plesek, J. Chem. ReV. 1992, 92, 269–278. (c) Yang, X.; King, W. A.; Sabat,
M.; Marks, T. J. Organometallics 1993, 12, 4254–4258. (d) Crowther, D. J.;
Swenson, D. C.; Jordan, R. F. J. Am. Chem. Soc. 1995, 117, 10403–10404.
(e) Hawthorne, M. F.; Zink, J. I.; Skelton, J. M.; Bayer, M. J.; Liu, C.;
Livshits, E.; Baer, R.; Neuhauser, D. Science 2004, 303, 1849–1851. (f)
Shen, H.; Xie, Z. Chem. Commun. 2009, 2431–2445. (g) Planas, J. G.;
Viñas, C.; Teixidor, F.; Comas-Vives, A.; Ujaque, G.; Lledós, A.; Light,
M. E.; Hursthouse, M. B. J. Am. Chem. Soc. 2005, 127, 15976–15982. (h)
Abram, P. D.; Ellis, D.; Rosair, G. M.; Welch, A. J. Chem. Commun. 2009,
5403–5405. (i) Armstrong, A. F.; Lebert, J. M.; Brennan, J. D.; Valliant,
J. F. Organometallics 2009, 28, 2986–2992. (j) Olejniczak, A. B.; Mucha,
P.; Gruner, B.; Lesnikowski, Z. L. Organometallics 2007, 26, 3272–3274.
(k) Cı́gler, P.; Kozı́sek, M.; Rezácová, P.; Brynda, J.; Otwinowski, Z.;
Pokorná, J.; Plesek, J.; Grüner, B.; Dolecková-Maresová, L.; Mása, M.;
Sedlácek, J.; Bodem, J.; Kräusslich, H.-J.; Král, V.; Konvalinka, J. Proc.
Natl. Acad. Sci. U.S.A. 2005, 102, 15394–15399. (l) Law, J. D.; Brewer,
K. N.; Herbst, R. S.; Todd, T. A.; Wood, D. J. Waste Management 1999,
19, 27–37.
(11) Hawthorne, M. F.; Dunks, G. B. Science 1972, 178, 462–471.
(12) Hamann, T. W.; Farha, O. K.; Hupp, J. T. J. Phys. Chem. C 2008, 112,
19756–19764.
(13) (a) Standridge, S. D.; Schatz, G. C.; Hupp, J. T. Langmuir 2009, 25, 2596–
2600. (b) Guo, J.; She, C.; Lian, T. J. Phys. Chem. C 2007, 111, 8979–
8987.
(14) Taguchi, T.; Zhang, X.-T.; Sutanto, I.; Tokuhiro, K.-I.; Rao, T. N.;
Watanabe, H.; Nakamori, T.; Uragami, M.; Fujishima, A. Chem. Commun.
2003, 2480–2481.
(15) (a) Palomares, E.; Clifford, J. N.; Haque, S. A.; Lutz, T.; Durrant, J. R.
J. Am. Chem. Soc. 2003, 125, 475–482. (b) Li, T. C.; Góes, M. S.; FabregatSantiago, F.; Bisquert, J.; Bueno, P. R.; Prasittichai, C.; Hupp, J. T.; Marks,
T. J. J. Phys. Chem. C 2009, 113, 18385–18390.
(16) Diamant, Y.; Chen, S. G.; Melamed, O.; Zaban, A. J. Phys. Chem. B 2003,
107, 1977–1981.
(17) (a) Fabregat-Santiago, F.; Garcı́a-Cañadas, J.; Palomares, E.; Clifford, J. N.;
Haque, S. A.; Durrant, J. R.; Garcia-Belmonte, G.; Bisquert, J. J. Appl.
Phys. 2004, 96, 6903–6907. (b) Palomares, E.; Clifford, J. N.; Haque, S. A.;
Lutz, T.; Durrant, J. R. Chem. Commun. 2002, 1464–1465.
(18) (a) O’Regan, B. C.; Scully, S.; Mayer, A. C.; Palomares, E.; Durrant, J. J.
Phys. Chem. B 2005, 109, 4616–4623. (b) Law, M.; Greene, L. E.;
Radenovic, A.; Kuykendall, T.; Liphardt, J.; Yang, P. J. Phys. Chem. B
2006, 110, 22652–22663.
(19) Zaban, A.; Greenshtein, M.; Bisquert, J. ChemPhysChem. 2003, 4, 859–
864.
(20) (a) Martinson, A. B. F.; Góes, M. S.; Fabregat-Santiago, F.; Bisquert, J.;
Pellin, M. J.; Hupp, J. T. J. Phys. Chem. A 2009, 113, 4015–4021. (b)
Martinson, A. B. F.; Elam, J. W.; Liu, J.; Hupp, J. T.; Pellin, M. J.; Marks,
T. J. Nano Lett. 2008, 8, 2862–2866.
(21) Clifford, J. N.; Palomares, E.; Nazeeruddin, M. K.; Grätzel, M.; Durrant,
J. R. J. Phys. Chem. C 2007, 111, 6561–6567.
(22) Peter, L. M. J. Phys. Chem. C 2007, 111, 6601–6612.
(23) (a) Wang, M.; Anghel, A. M.; Marsan, B.; Ha, N.-L.; Pootrakulchote, N.;
Zakeeruddin, S. M.; Grätzel, M. J. Am. Chem. Soc. 2009, 131, 15976–
15977. (b) Bay, L.; West, K.; Winther-Jensen, B.; Jacobsen, T. Sol. Energy
Mater. Sol. Cells 2006, 90, 341–351.
JA100396N
�
 Alexander Spokoyny
Alexander Spokoyny