

Abstract
Material development involves laborious processes to explore the vast materials space. The key to accelerating these processes is understanding the structure-functionality relationships of materials. Machine learning has enabled large-scale analysis of underlying relationships between materials via their vector representations, or embeddings. However, the learning of material embeddings spanning most known inorganic materials has remained largely unexplored due to the expert knowledge and efforts required to annotate large-scale materials data. Here we show that our self-supervised deep learning approach can successfully learn material embeddings from crystal structures of over 120â000 materials, without any annotations, to capture the structure-functionality relationships among materials. These embeddings revealed the profound similarity between materials, or 'materials concepts', such as cuprate superconductors and lithium-ion battery materials from the unannotated structural data. Consequently, our results enable us to both draw a large-scale map of the materials space, capturing various materials concepts, and measure the functionality-aware similarities between materials. Our findings will enable more strategic approaches to material development.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The diverse properties of the inorganic materials originate from their crystal structures, i.e. the atomic-scale periodic arrangements of elements. How structures determine low-level material properties such as the band gap and formation energy is well studied as the structure-property relationship [1, 2]. On the other hand, the materials science literature often discusses 'superconductors' [3], 'permanent magnets' [4], or 'battery materials' [5], referring to their higher-level properties, or functionality. Nevertheless, understanding what structures exhibit such functionality, or understanding the structure-functionality relationship, is a fundamental question in materials science. We call this functionality-level material similarity 'materials concepts'. Traditionally, materials science has sought new materials by experimentally and theoretically understanding specific functionalities of materials in a bottom-up fashion [1â5]. However, this labour-intensive narrowly focused analysis has prevented us from grasping the whole picture of the materials space across various materials concepts. For next-generation material discovery based on the structure-functionality relationship, we argue here the need for a top-down unified view of crystal structures through materials concepts. We pursue this ambition by learning a latent representation space of crystal structures. Thus, this representation space should ideally both (a) recognise materials concepts at scale and (b) be equipped with a functionality-level similarity metric between materials. We here utilise multi-modal structural attributes of materials to effectively capture structural patterns correlated to material functionality (figure 1). The underlying hypothesis here is that materials concepts are the intrinsic nature of crystal structures, and therefore, deeply analysing the structural similarity between materials will lead to capturing functionality-level similarity.
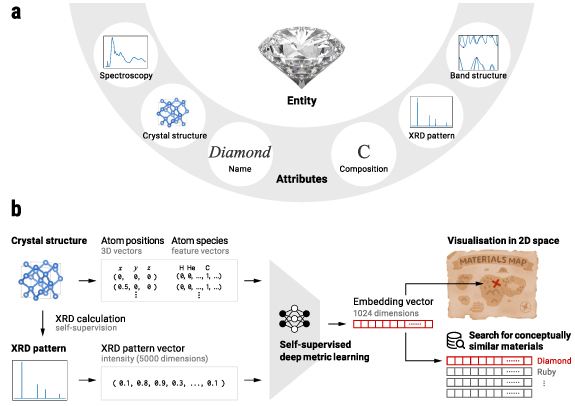
Figure 1. Our strategy for learning materials concepts. (a) Diamond as an example material and its various structural attributes showing different information of diamond in different data forms, or modalities. Since each attribute has its own advantages and disadvantages in expressing a material, using multiple attributes for a material can provide a more comprehensive view of the material. Particularly, the combination of the crystal structure and the x-ray diffraction (XRD) pattern, which we employ in this study, is known to well reflect two complementary structural features of materials, the local structure and the periodicity [1]. (b) Our goal is to represent each material as an abstract constant size vector (embedding) whose distances to other embeddings reflect conceptual (functionality-level) similarities between materials. These embeddings allow us to visualise the materials space intuitively and also to search for conceptually similar materials given a query material. We learn embeddings from pairs of crystal structures and XRD patterns in the framework of deep metric learning. This cross-modal learning approach trains deep neural networks by teaching them that each pair should represent the same material entity. Because the XRD pattern can be theoretically calculated from the crystal structure, this learning can be performed in a self-supervised manner without any explicit human annotations for the materials dataset.
Download figure:
Standard image High-resolution imageFigure 3(a) highlights key results by our representation space, which maps the crystal structures of materials to abstract 1024-dimensional vectors. For visualisation, these vectors were reduced to 2D plots in the figure using a dimensionality reduction technique called t-distributed stochastic neighbour embedding (t-SNE) [6]. We target 122â543 inorganic materials registered in the Materials Project (MP) database (amounting to 93%) to capture nearly the entire space of practically known inorganic materials. These crystal structures themselves contain information about their functionalities implicitly. However, they do not explicitly tell us what structural patterns lead to specific material functionalities such as superconductivity due to complicated structure-functionality relationships. Nevertheless, these materials form clusters of various materials concepts in the space (see annotations in figure 3(a)), showing the success of our representation space capturing structural patterns correlated to material functionality.
When analysing diverse relationships entangled with complex features in large-scale data, machine learning (ML) and deep neural networks (DNNs) are key technologies [7, 8]. Indeed, these technologies often surpass human ability in recent materials informatics work. For example, when extracting features from materials data for complex tasks such as physical property prediction, learning-based descriptors [9â18] have been shown to outperform traditional hand-crafted descriptors [19â24].
Likewise, representation learning is gaining attention for understanding human-incomprehensible large-scale materials data [25â29], visualising the materials space [25, 27â30], and generating crystal structures [31â36]. These material representations aim to map the abstract, comprehensive information of each material into a vector called an 'embedding'. Our work has the same purpose as that of embedding learning. However, to date, neither descriptor nor embedding learning explicitly learns the underlying relationships or similarities between materials. Particularly, existing embeddings [25â29] are learned indirectly as latent feature vectors in an internal layer of a DNN by addressing a surrogate training task (e.g. the prediction of physical properties [27, 28], a task of natural language processing (NLP)[26] or its variants [25, 29]). In such an approach, it is unclear from which layer we should obtain the latent vectors or which metric we should use to measure the distance/similarity between them.
Capturing abstract concepts of materials via learning structural similarities between them is analogous to word embedding learning [26] in NLP. Similar to materials concepts, meanings of words in natural languages often reside in complex and abstract notions, which prevent us from acquiring precise definitions for them. Word embeddings then attempt to capture individual word concepts, without being explicitly taught, by absorbing our word notions implicitly conveyed in the contexts provided by a large-scale text corpus. Once optimised, similarities/distances between embeddings express their concepts, e.g. the embedding of 'apple' will be closer to those of other fruits such as 'grape' and 'banana' than 'dog' or 'cat'. Our crystal structure embedding shares a similar spirit with word embedding in that both attempt to capture abstract concepts via learned similarities. More importantly, we exploit a large-scale material database as a corpus of materials that implicitly conveys important structural patterns in its contexts of crystal structures, as analogously to word embedding. These structure instances of diverse kinds of materials, even without explicit annotations about their properties, should contain tacit but meaningful information about physics and material functionality that can guide the learning of ML models. From an ML perspective, such a learning strategy is called self-supervised learning [37], in which the data of interest themselves provide supervision.
In this study, we demonstrate the large-scale self-supervised learning of material embeddings using DNNs. In essence, we follow the principle that the structure determines properties and aim to discover materials concepts purely from crystal structures without explicit human supervision in learning. To this end, we use a collection of crystal structures as the only source of training data and do not provide any annotation regarding specific material properties (e.g. class labels such as 'superconductors' and 'magnets', or property values such as superconducting transition temperature and magnetisation). Furthermore, unlike existing methods for material embedding learning, we explicitly optimise the relationships between embeddings by pioneering the use of deep metric learning [38]. Metric learning is an ML framework for learning a measure of similarity between data points. Unlike the common practice of metric learning performed in a supervised fashion using annotated training data [38], we allow our ML model to be learned from the unannotated structural data in a self-supervised fashion.
2. Results
Our key idea for self-supervised learning, illustrated in figure 2(a), is to learn unified embedding representations for paired inputs expressing two complementary structural features characterising materials: the local structure and the periodicity [1]. In our model, the local structure is represented by a graph whose nodes and edges stand for the atoms and their connections. The periodicity is represented by a simulated x-ray diffraction (XRD) pattern, which can be theoretically calculated from the crystal structure using Bragg's law and Fourier transformation [1]. We simultaneously train two DNN encoders by enforcing them to produce consistent embeddings across the two different input forms. This training strategy follows a simple optimisation principle: (a) for a positive pair in which the input crystal structure and XRD pattern come from the same material, the Euclidean distance between two embedding vectors is decreased, and (b) for a negative pair in which these inputs come from different materials, the distance is increased. We implement this principle in the form of a bidirectional triplet loss function, as illustrated in figure 2(b). For the detailed method protocol, see section 5.
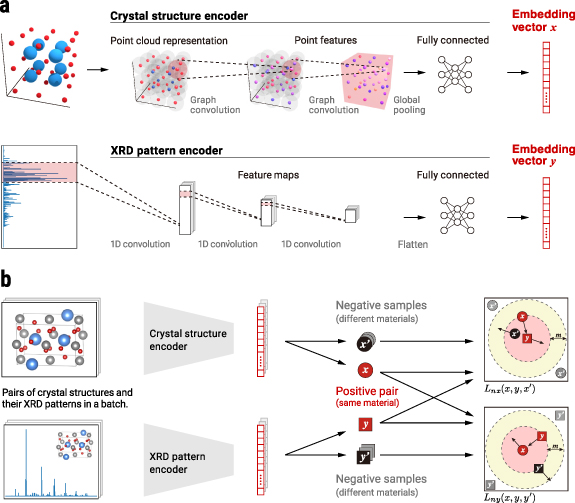
Figure 2. Our self-supervised deep metric learning for materials. (a) The proposed network architecture, including two separate DNNs to encode the crystal structures and XRD patterns into unified embedding vector representations. To account for respective input data forms (figure 1(b)), the crystal-structure encoder employs a DNN for graphs while the XRD pattern encoder employs a 1D convolutional neural network. (b) A schematic view of our bidirectional triplet loss. This triplet loss is used to simultaneously train the two DNN encoders to output embeddings that are close together when the input crystal structures and XRD patterns are paired (red-coloured x and y ) and far from one another when the inputs are not paired ( x and y vs others). More details are given in section 5.
Download figure:
Standard image High-resolution image
Figure 3. A map of the materials space. (a) A global map of the materials space, plotted via a t-SNE visualisation of the embeddings. Each point corresponds to an individual material that is encoded by the trained crystal-structure encoder. The map was annotated with cluster labels through manual inspection. Our materials map is publicly accessible via an interactive website (https://github.com/quantumbeam/materials-concept-learning). (b) A close-up view of a cluster of cuprate superconductors in the materials space. (c) Crystal structures of Y-123 and Y-124 families, which are closely distributed in (b). The CuO chain in Y-123 and the double CuO chain in Y-124, which is similar to vertically repeating Y-123, are important features of YBCO superconductors.
Download figure:
Standard image High-resolution imageBy design, we minimise human knowledge of specific materials concepts in both the data source and training process, with the belief that materials concepts are buried in crystal structures. This design principle enhances the significance of the resulting embedding highlighted earlier (figure 3(a)). It captures profound materials relationships through simple data and optimisation operations considering only general and elementary knowledge of materials such as crystallographic data and Bragg's law. The results suggest that materials concepts can be exposed in deeply-transformed abstract expressions unifying the complementary factors, i.e. the local structure and periodicity, of crystal structures.
The following analyses examine the embedding characteristics more carefully to see if the embedding space has the two desired features mentioned above. Specifically, we qualitatively analyse (a) the global embedding distribution using t-SNE visualisation and (b) the local neighbourhoods around some important materials using the learned similarity metric between crystal structures. In the latter, a superconductor (Hg-1223), a lithium-ion battery material (LiCoO2), and some magnetic materials serve as our benchmark materials because of the high social impacts and the diverse properties yet complex structures of these material classes. These analyses also demonstrate the usefulness of our materials map visualisation and similarity metric for material discovery and development.
2.1. Global distribution analysis
Careful inspection of the embedding space (figure 3(a)) reveals various clusters consistent with our knowledge of materials. Here, we note several interesting examples. A series of clusters corresponding to double perovskites (A2BBʹX6) with different anions, X, exists along the left edge and at the centre of the map, forming a family of materials with the same prototypical crystal structure. This layout suggests that our model captures the structural similarity while properly distinguishing the local atomic environment at each site. At the lower left of the map, well-known 2D materials (transition metal dichalcogenides) form clusters in accordance with their atomic stacking structures [39]. At the top edge lies a cluster of imaginary unstable materials with extremely low-density structures (see also figure 5(a) for more details), representing one of the simplest cases of crystal structures governing physical properties. This cluster of unstable materials is an example showing that our embeddings capture materials characteristics solely from crystal structures without any explicit annotation given for training.
One exciting finding from this map is a cluster of cuprate superconductors at the left edge. This cluster includes the first-discovered copper oxide superconductor, the LaâBaâCuâO system, and the well-known high-transition-temperature ( ) superconductors YBCO (YBa2Cu3O7 or Y-123), which are located close to LaâBaâCuâO. These celebrated superconductors share a common structural feature, a CuO2 plane, that is vital to their superconductivity [3]. The formation of this cluster suggests that our embeddings recognise this hallmark structural feature. A closer look at this cluster (figure 3(b)) further reveals the presence of subclusters with structural features linking them. Y-123 and its variant Y-124 have a non-trivial structural similarity related to the CuO chain (see figure 3(c)). In addition, we confirmed other major cuprate superconductors containing Bi, Tl, Pb, or Hg form respective clusters in accordance with their local structures called 'block layers', a key structural concept for understanding the underlying physics of cuprate superconductors [40]. The proximity of these materials on the map further supports the claim that the embeddings capture the structural characteristics and, consequently, the structure-functionality relationships between cuprate superconductors.
) superconductors YBCO (YBa2Cu3O7 or Y-123), which are located close to LaâBaâCuâO. These celebrated superconductors share a common structural feature, a CuO2 plane, that is vital to their superconductivity [3]. The formation of this cluster suggests that our embeddings recognise this hallmark structural feature. A closer look at this cluster (figure 3(b)) further reveals the presence of subclusters with structural features linking them. Y-123 and its variant Y-124 have a non-trivial structural similarity related to the CuO chain (see figure 3(c)). In addition, we confirmed other major cuprate superconductors containing Bi, Tl, Pb, or Hg form respective clusters in accordance with their local structures called 'block layers', a key structural concept for understanding the underlying physics of cuprate superconductors [40]. The proximity of these materials on the map further supports the claim that the embeddings capture the structural characteristics and, consequently, the structure-functionality relationships between cuprate superconductors.
These findings naturally lead us to the idea that the map might be able to identify potential superconductors or other beneficial compounds that have not yet been recognised. We leave this idea as an open question and have set up a project website where anyone can dig into the embedding map to search for, or rediscover, potential compounds with preferable functionality.
The t-SNE visualisation also provides a macroscopic perspective on the materials space based on the crystal structure. The simplest indicator of success for this model is the distribution of the elements within the materials map. Because atoms and ions with similar electron configurations compose materials with the same or similar crystal structures, we expect the element distributions to show cluster-like features if our embeddings have been trained successfully. In figure 4, we highlight each element in the map and display all elements in the form of a periodic table. As expected, figure 4 clearly shows similar distributions of blue-coloured clusters in the vertical and horizontal directions. These distributions can be analogously called the 'alkali metal plateau', the '3d transition metal district', or 'rare-earth mountains' if we follow the map metaphor, indicating that the embeddings succeed in capturing the similarities of roles between elements in crystal structures. Additionally, we noticed that well-known connections between physical properties and elements can also be probed using this plotting technique (see figures 5(b) and (c) for details). Although these visualisations (figures 4 and 5) are intended to confirm expected outcomes rather than showing interesting findings, they demonstrate their potential utility, e.g. for giving researchers new insights or helping them find materials with desired properties.

Figure 4. Elemental distributions within the material embedding space. t-SNE plots of the embeddings are laid out on the periodic table, coloured blue or grey according to whether each material contains the corresponding element or not. Similar distributions in the vertical and horizontal directions (groups and periods) of the table indicate that the embeddings successfully capture the similarities of roles between elements in crystal structures. 'n.a.' means no material containing the element is found in our dataset.
Download figure:
Standard image High-resolution image
Figure 5. Physical property distributions within the material embedding space. t-SNE plots of the embeddings are coloured according to the physical properties: energy above the hull, band gap, and magnetisation. These plots show clusters of materials with similar physical properties, indicating that the embeddings capture the property similarities between materials. (a) The distribution of energy above the hull (eV). A large value of energy above the hull indicates that a material is unstable. A cluster of unstable compounds containing sparse unsynthesisable crystal structures was identified on the upper left. (b) The distribution of the band gap (eV). The distribution overlap of large-bandgap materials in this figure and oxides in figure 4 demonstrates a well-known connection between the band gap and oxygen. (c) The distribution of magnetisation (T). Materials with large magnetic moments have higher composition ratios of magnetic elements such as Mn, Fe, Co, and Ni and are particularly studied in the rare-earth permanent magnet research. The distinct yellow cluster in the top right of this figure contains intermetallic compounds of the magnetic elements and rare-earth elements (e.g. Ce, Pr, Nd, and Sm), as evident from figure 2 where the distributions of these elements overlap in this area.
Download figure:
Standard image High-resolution image2.2. Local neighbourhood analysis
We next examine the local neighbourhoods of several benchmark areas to verify whether the learned metric recognises functionality-level material similarity. Since the embeddings were optimised with the Euclidean distance, we also used this metric to determine the neighbourhoods.
As the first example, we analysed the neighbourhoods of Hg-1223, a superconductor with the highest known  (134âK) at ambient pressure [42]. To our surprise, the first and second nearest neighbourhoods correspond to its close kin Hg-1234 and Hg-1212, which also have high
(134âK) at ambient pressure [42]. To our surprise, the first and second nearest neighbourhoods correspond to its close kin Hg-1234 and Hg-1212, which also have high  values (125âK and 90âK) but different block layers [40] from those of Hg-1223 (see figure 6(a)). Further investigation identified major Tl-based high-
values (125âK and 90âK) but different block layers [40] from those of Hg-1223 (see figure 6(a)). Further investigation identified major Tl-based high- superconductors, such as Tl-2234 (
superconductors, such as Tl-2234 ( = 112âK), Tl-2212 (
= 112âK), Tl-2212 ( = 108âK), and Tl-1234 (
= 108âK), and Tl-1234 ( = 123âK)[43], and many other superconductors occupying the top-50 neighbourhoods (see table 1). The connection between the crystal structures and
= 123âK)[43], and many other superconductors occupying the top-50 neighbourhoods (see table 1). The connection between the crystal structures and  values involves non-trivial mechanisms that are not immediately evident from the crystal structures [3, 40]. The results suggest that our model effectively bridges this gap with the help of learned structure-functionality relationships that are deeply buried in the 1024-dimensional space.
values involves non-trivial mechanisms that are not immediately evident from the crystal structures [3, 40]. The results suggest that our model effectively bridges this gap with the help of learned structure-functionality relationships that are deeply buried in the 1024-dimensional space.
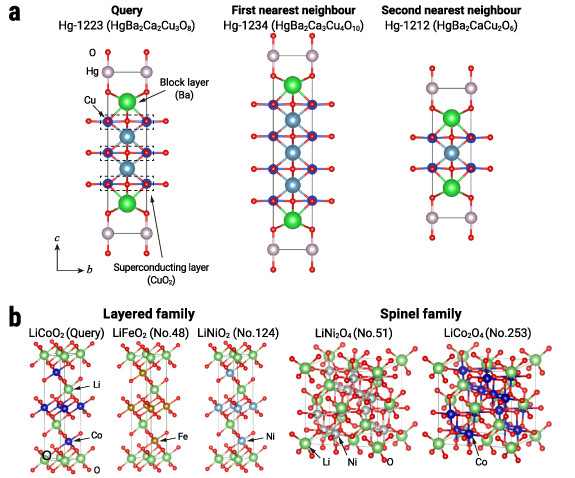
Figure 6. Crystal structures of queries and nearest neighbours. (a) Crystal structures of the Hg-1223 superconductor and its first and second nearest neighbours, Hg-1234 and Hg-1212, in the embedding space. These are all Hg-based copper oxide superconductors with high  and are built on two important components, namely, block layers and superconducting layers. Placing these close-kin materials, yet with different numbers of superconducting layers, as close neighbours suggests that the embedding captures the conceptual similarity in their structures. See also table 1 for the top-50 neighbour list of Hg-1223, in which more superconductors with high
and are built on two important components, namely, block layers and superconducting layers. Placing these close-kin materials, yet with different numbers of superconducting layers, as close neighbours suggests that the embedding captures the conceptual similarity in their structures. See also table 1 for the top-50 neighbour list of Hg-1223, in which more superconductors with high  are found. (b) Crystal structures of the well-known lithium-ion battery cathode material LiCoO2 and its neighbours in the embedding space. The layered and spinel families, which are two major types of the cathode materials, were identified in the neighbourhoods. See also table 2 for the list of the top-50 neighbours, in which more layered-family materials are found.
are found. (b) Crystal structures of the well-known lithium-ion battery cathode material LiCoO2 and its neighbours in the embedding space. The layered and spinel families, which are two major types of the cathode materials, were identified in the neighbourhoods. See also table 2 for the list of the top-50 neighbours, in which more layered-family materials are found.
Download figure:
Standard image High-resolution imageTable 1. The top-50 neighbours of Hg-1223 in comparison with hand-crafted descriptors.
| Â | Our embedding | Ewald sum matrix | Sine Coulomb matrix | |||
|---|---|---|---|---|---|---|
| No. | Formula | ID | Formula | ID | Formula | ID |
| Query | Ba2Ca2Cu3HgO8 | mp-22601 | Ba2Ca2Cu3HgO8 | mp-22601 | Ba2Ca2Cu3HgO8 | mp-22601 |
| 1 | Ba2Ca3Cu4HgO10 | mp-1228579 | Sr4TlFe2O9 | mp-1218464 | Tl(CuTe)2 | mp-569204 |
| 2 | Ba2CaCu2HgO6 | mp-6879 | Ba2La2Ti2Cu2O11 | mp-1214655 | CaLa2BiO6 | mvc-15176 |
| 3 | Ba6Ca6Cu9Hg3O25 | mp-1228760 | CeY4Mg5 | mp-1226574 | PtC4S2(IO)2 | mp-1102535 |
| 4 | Sr2CaCu2(BiO4)2 | mp-1218930 | Ba6Nb2Ir(ClO6)2 | mp-558113 | Ba2FeReO6 | mp-31756 |
| 5 | Ba10Ca5Cu10Hg5O31 | mp-1229139 | Ba6Ru2Pt(ClO6)2 | mp-554949 | Hg(SbO3)2 | mp-754065 |
| 6 | SrCa2Cu2(BiO4)2 | mp-1208800 | Ba2Nd2Ti2Cu2O11 | mp-557043 | Ba2CuWO6 | mp-505618 |
| 7 | Ba8Ca4Cu8Hg4O25 | mp-1228371 | Ba4ScTi4BiO15 | mp-1228157 | CaLa2WO6 | mvc-15479 |
| 8 | Ba2Ca3Tl2(CuO3)4 | mp-556574 | La3ZnNi3 | mp-18573 | Ba2YTaO6 | mp-12385 |
| 9 | Ba2Mg3Tl2(WO3)4 | mvc-129 | Zr4WC5 | mp-1215364 | TlCdTe2 | mp-998919 |
| 10 | Ba2TlV2O7 | mvc-2978 | Nd3GaCo3 | mp-1103877 | TlCuPd2 | mp-1096374 |
| 11 | Sr2YCu2(BiO4)2 | mp-1208863 | Y4Ti6Bi2O21 | mp-1216208 | LaTlAg2 | mp-867817 |
| 12 | Sr2LaCu2HgO6 | mp-1208803 | Sm3HoS4 | mp-1219190 | In3Au | mp-973498 |
| 13 | Ba2CaTl2(CuO4)2 | mp-573069 | Ba3Bi(BO2)9 | mp-1200141 | CeTlAg2 | mp-867298 |
| 14 | Ba4CaCu6(HgO8)2 | mvc-15237 | AgRhO2 | mp-997106 | Cs2WBr6 | mp-541753 |
| 15 | Ba4Ca4Cu6Hg2O17 | mp-1228265 | YbSm3S4 | mp-1215523 | TlIn3 | mp-1187742 |
| 16 | Ba2AlTlCo2O7 | mvc-2977 | Ca4Cd3Au | mp-1227562 | In3Pt | mp-1184857 |
| 17 | Sr8Pr4Cu9(HgO8)3 | mp-1218674 | InAg4 | mp-1223819 | Ca4Cd3Au | mp-1227562 |
| 18 | Ba6Ca3Cu6Hg3O19 | mp-1228161 | Sr4ZrTi3O12 | mp-1218457 | Cd3Pt | mp-1183641 |
| 19 | Ba8Ca8Tl7(Cu4O13)3 | mp-1204270 | Ce3Ni2Ge7 | mp-1213875 | Ag3Au | mp-1183214 |
| 20 | Ba4Ca4Tl3Cu6O19 | mp-542197 | Ba2YTlV2O7 | mvc-2994 | Mn4BiSb3 | mp-1221739 |
| 21 | Ba6Ca6Tl5Cu9O29 | mp-680433 | Te3Au | mp-1217358 | NdTlAg2 | mp-974782 |
| 22 | Ba2AlTlCo2O7 | mp-1266279 | Nd3Cu4(P2O)2 | mp-1209832 | HgI3 | mp-973601 |
| 23 | Ba2Ca2Tl2Ni3O10 | mvc-3067 | Ba4Zn4B14Pb2O31 | mp-1194514 | TlCdIn2 | mp-1093975 |
| 24 | Ba2Ca2Tl2Cu3O10 | mp-653154 | Ba6Na2Nb2P2O17 | mp-556637 | CePd2Pt | mp-1226474 |
| 25 | Ba2Ca2Tl2Co3O10 | mvc-3021 | Ba2Tb2Ti2Cu2O11 | mp-505223 | PmHgRh2 | mp-862913 |
| 26 | Sr2CaCu2(BiO4)2 | mp-555855 | Sc2TlCu3S5 | mp-1209018 | Sr2LaCu2HgO6 | mp-1208803 |
| 27 | Ba4Tl2Cu2HgO10 | mp-561182 | Eu(GaGe2)2 | mp-1225812 | NdPd2Pb | mp-1186317 |
| 28 | Ba6Ca12Cu15Hg3O37 | mp-1229082 | AgTe3 | mp-1229041 | PmTlRh2 | mp-862967 |
| 29 | BaCuReO5 | mvc-7248 | Sm3GaCo3 | mp-1105102 | Cd3Ir | mp-1183645 |
| 30 | Ba2Ca3Tl2(FeO3)4 | mvc-145 | Nb4Rh | mp-1220441 | HgPd3 | mp-1184658 |
| 31 | Sr10Cu5Bi10O29 | mp-667638 | La3(Al2Si3)2 | mp-1211155 | SnPd2Au | mp-1095757 |
| 32 | Ba2Ca3TlCu4O11 | mp-1228589 | Ce2In8Pt | mp-1103614 | PmTlAg2 | mp-862966 |
| 33 | Ba2Ca3Tl2(CuO3)4 | mp-556733 | CaNb2Bi2O9 | mp-555616 | Rb2LaAuCl6 | mp-1113498 |
| 34 | La2B3Br | mp-568985 | Ce2In8Ir | mp-1207157 | VAg3HgO4 | mp-1216423 |
| 35 | BaTl(SbO3)2 | mvc-10727 | Tc6BiO18 | mp-1101632 | In2SnHg | mp-1097125 |
| 36 | Sr10Cu5Bi10O29 | mp-652781 | Sb3Au | mp-1219474 | PrBiPd2 | mp-976884 |
| 37 | Ba2Tl2Zn2Cr3O10 | mvc-3164 | Sr4LaCl11 | mp-1218463 | TlIn3 | mp-1216611 |
| 38 | Ba2Ca2Tl2Fe3O10 | mvc-3027 | LaBiS2O | mp-1078328 | Cd2AgPt | mp-1096169 |
| 39 | Ba2Ti3Tl2O10 | mvc-2939 | HfNb4CN4 | mp-1224363 | Rb2CeAuCl6 | mp-1113397 |
| 40 | Sr2TaAlCu2O7 | mp-1251503 | MoN | mp-1078389 | In2SnPb | mp-1223808 |
| 41 | Ba2Mg3Tl2(SnO3)4 | mvc-10576 | YZnGe | mp-13160 | Cd2AgPt | mp-1183537 |
| 42 | Sr2AlTlCo2O7 | mp-1252241 | Pr3(Al2Si3)2 | mp-571302 | Ag2PdAu | mp-1096329 |
| 43 | Ba2AlTlV2O7 | mp-1265780 | Sr2(BiPd)3 | mp-1207133 | Ag3AuS2 | mp-34982 |
| 44 | Ba2CaTl2(CuO4)2 | mp-6885 | Na3HoTi2Nb2O12 | mp-676988 | PmRh2Pb | mp-862958 |
| 45 | Sr2LaCu2(BiO4)2 | mp-1209034 | Sr2YCu2BiO7 | mvc-280 | PmPd2Pb | mp-862950 |
| 46 | Ba2AlTlV2O7 | mvc-3002 | Na3DyTi2Nb2O12 | mp-689927 | Sr2PrTlCu2O7 | mp-1208792 |
| 47 | Ba2Mg3Tl2(FeO3)4 | mvc-28 | Rb3NaRe2O9 | mp-1209462 | InAg2Au | mp-1093943 |
| 48 | Sr2DyCu2(BiO4)2 | mp-1209149 | Sr3Fe2Ag2S2O5 | mp-1208725 | Ag2SnBiS4 | mp-1229127 |
| 49 | Ba2CuHgO4 | mp-6562 | Ba2Pr(CuO2)3 | mp-1228546 | Sb3Au | mp-29665 |
| 50 | Ba2Tl2W3O10 | mvc-3144 | Ce3(Al2Si3)2 | mp-29113 | PmAg2Pb | mp-862876 |
We compare the top-50 neighbours of the Hg-1223 superconductor obtained by using our embedding and two hand-crafted descriptors (Ewald sum matrix and sine Coulomb matrix) [22]. The query material, Hg-1223 (HgBa2Ca2Cu3O8), has the highest known  (134âK) at ambient pressure. Quite impressively, the neighbour list obtained by our embedding seems to be completely filled with superconductors, including the well-known Hg-1224 (No. 1) and Hg-1212 (No. 2) as well as Tl-based high-
(134âK) at ambient pressure. Quite impressively, the neighbour list obtained by our embedding seems to be completely filled with superconductors, including the well-known Hg-1224 (No. 1) and Hg-1212 (No. 2) as well as Tl-based high- superconductors such as Tl-2234 (No. 8), Tl-1234 (No. 32), and Tl-2212 (No. 44). By contrast, the lists obtained by the two existing descriptors contain irrelevant materials rather than superconductors. These results clearly show that our approach captures the conceptual similarity between superconductors, which is undetectable by the existing descriptors. See also the SI (appendix A3) for the detailed procedures of the descriptor computations and more discussions.
superconductors such as Tl-2234 (No. 8), Tl-1234 (No. 32), and Tl-2212 (No. 44). By contrast, the lists obtained by the two existing descriptors contain irrelevant materials rather than superconductors. These results clearly show that our approach captures the conceptual similarity between superconductors, which is undetectable by the existing descriptors. See also the SI (appendix A3) for the detailed procedures of the descriptor computations and more discussions.
Table 2. The top-50 neighbours of LiCoO2 in comparison with hand-crafted descriptors.
| Â | Our embedding | Ewald sum matrix | Sine Coulomb matrix | |||
|---|---|---|---|---|---|---|
| No. | Formula | ID | Formula | ID | Formula | ID |
| Query | LiCoO2 | mp-22526 | LiCoO2 | mp-22526 | LiCoO2 | mp-22526 |
| 1 | Li14MgCo13O28 | mp-769537 | LiNiO2 | mp-25587 | LiCoO2 | mp-1222334 |
| 2 | Li4Co3NiO8 | mp-867537 | Co(HO)2 | mp-24105 | CoHO2 | mp-27913 |
| 3 | Li3Fe(CoO3)2 | mp-761602 | LiFeO2 | mp-1222302 | LiCoF2 | mp-1097040 |
| 4 | Li3(CoO2)4 | mp-850808 | LiNiO2 | mp-25316 | LiCoN | mp-1246462 |
| 5 | Li3MnCo3O8 | mp-774219 | Li2NiO2 | mp-19183 | Li2CoN2 | mp-1247124 |
| 6 | Li20(CoO2)21 | mp-532301 | MgMnN2 | mp-1247154 | Be5Co | mp-1071690 |
| 7 | Li3CrCo3O8 | mp-849768 | Li2CaCd | mp-1096283 | Be3Co | mp-1183423 |
| 8 | Li3MnCo3O8 | mp-758163 | NiO2 | mp-25210 | Be2Co | mp-1227342 |
| 9 | Li8FeCo9O20 | mp-764865 | LiFeOF | mp-775022 | CoCN | mp-1245659 |
| 10 | Li3Co2NiO6 | mp-765538 | MnO2 | mp-1221542 | Li3Co | mp-976017 |
| 11 | Li3CrCo3O8 | mp-759149 | Co(HO)2 | mp-625939 | Li2CoO2 | mp-755133 |
| 12 | Li3TiCo3O8 | mp-757214 | Co(HO)2 | mp-625943 | Li2CoO2 | mp-755297 |
| 13 | Li4MgCo3O8 | mp-754576 | Li2CuO2 | mp-1239022 | Be12Co | mp-1104193 |
| 14 | Li5Co2Ni3O10 | mp-769553 | CoO2 | mp-1062939 | CoO2 | mp-1181781 |
| 15 | Li(CoO2)2 | mp-552024 | NaCoO2 | mp-1221066 | CoO2 | mvc-13108 |
| 16 | Li14Co13O28 | mp-777836 | NiO2 | mp-634706 | Co(HO)2 | mp-626708 |
| 17 | Li3(NiO2)5 | mp-762165 | MgMnO2 | mp-1080243 | Co(HO)2 | mp-625939 |
| 18 | Li2CoO2F | mp-764063 | LiCuF2 | mp-753098 | Co(HO)2 | mp-625943 |
| 19 | Li2(CoO2)3 | mp-758539 | Ni(HO)2 | mp-625074 | Co(HO)2 | mp-24105 |
| 20 | Li5Fe2Co3O10 | mp-769566 | CrO2 | mp-1009555 | CoO2 | mp-1062939 |
| 21 | Li2CoNi3O8 | mp-752703 | CoHO2 | mp-27913 | CoO2 | mp-1062643 |
| 22 | Li10Fe3Co7O20 | mp-760848 | NaLi2As | mp-1014873 | CoO2 | mp-556750 |
| 23 | Li7Co5O12 | mp-771155 | LiNiO2 | mp-25411 | CoH3 | mp-1183678 |
| 24 | Li3(NiO2)4 | mp-755972 | Li2CoO2 | mp-755133 | CoH | mp-1206874 |
| 25 | Li9Ni15O28 | mp-759153 | LiCuO2 | mp-754912 | CoO2 | mp-1063268 |
| 26 | Li20Co21O40 | mp-685270 | CrN2 | mp-1014264 | CoN | mp-1008985 |
| 27 | Li7(NiO2)11 | mp-768079 | MgCr | mp-973060 | CoN | mp-1009078 |
| 28 | Li2(NiO2)3 | mp-762391 | Ni(HO)2 | mp-1180084 | FeHO2 | mp-755285 |
| 29 | Li4Co2Ni3O10 | mp-778996 | Co(HO)2 | mp-626708 | LiFeO2 | mp-1222302 |
| 30 | Li2Co3NiO8 | mp-757851 | Ni(HO)2 | mp-27912 | LiFeO2 | mp-19419 |
| 31 | LiCoNiO4 | mp-754509 | VO | mp-19184 | CoBO3 | mp-1183397 |
| 32 | Li4(NiO2)7 | mp-774600 | Be4AlFe | mp-1227272 | LiFeOF | mp-775022 |
| 33 | Li(CoO2)2 | mp-774082 | FeO2 | mp-1062652 | LiNiO2 | mp-25411 |
| 34 | Li(CoO2)2 | mp-752807 | LiCoF2 | mp-1097040 | NiHO2 | mp-1067482 |
| 35 | Li8(NiO2)11 | mp-758772 | LiFeO2 | mp-19419 | Li4Co(OF)2 | mp-850355 |
| 36 | Li3CoNi3O8 | mp-774300 | Na2NiO2 | mp-752558 | NiHO2 | mp-999337 |
| 37 | Li2CoNi3O8 | mp-1178042 | Li2CuO2 | mp-4711 | LiNiO2 | mp-25587 |
| 38 | Li7(NiO2)8 | mp-690528 | Li2CoO2 | mp-755297 | LiNiO2 | mp-25316 |
| 39 | Li10Co3Ni7O20 | mp-769555 | MgCr | mp-1185858 | LiFeO3 | mp-1185320 |
| 40 | Li7Ni13O24 | mp-758593 | Sc2CO | mp-1219429 | LiFeN | mp-1245817 |
| 41 | Li9Co7O16 | mp-1175506 | MnBO3 | mp-1185996 | CoNF3 | mp-1213745 |
| 42 | Li3Cr(CoO3)2 | mp-761831 | VN | mp-1001826 | LiNiO3 | mp-1185261 |
| 43 | Li2Co3NiO8 | mp-778768 | NiHO2 | mp-999337 | Li4FeN2 | mp-28637 |
| 44 | Li2FeCo3O8 | mp-1177976 | CrO | mp-19091 | LiNiN | mp-29719 |
| 45 | Li4Co3(NiO4)3 | mp-777850 | Ni(HO)2 | mp-625072 | Be3Fe | mp-983590 |
| 46 | Li3Al2CoO6 | mp-1222591 | VN | mp-925 | NiO3 | mp-1209929 |
| 47 | Li(NiO2)2 | mp-752531 | GaH6N2F3 | mp-1224894 | Be5Fe | mp-1025010 |
| 48 | LiFeO2 | mp-19419 | Fe(HO)2 | mp-626680 | Li2FeO2 | mp-755094 |
| 49 | Li4AlNi3O8 | mp-1222534 | CrN | mp-1018157 | Be12Fe | mp-1104104 |
| 50 | Li3CoNi3O8 | mp-757871 | VN | mp-1018027 | FeB2 | mp-569376 |
We compare the top-50 neighbours of LiCoO2 obtained by using our embedding and two hand-crafted descriptors (Ewald sum matrix and sine Coulomb matrix) [22]. The query material, LiCoO2, is one of the most crucial lithium-ion battery cathodes. In the list of our embedding, the many neighbours of LiCoO2 are occupied by LiCo Mx
O2 families with the same layered structure as LiCoO2 but partly substituted with different transition metals M. Since these partial substitutions are represented as supercells, the system's apparent size is larger than the original unit cells. Our approach is unaffected by these apparent differences and can recognise the essential similarities. While most of our list is filled with lithium oxides, the other two lists obtained by the existing descriptors do not suggest this consistent trend. These results suggest that our model recognises the concept of lithium-ion battery cathodes, which is not captured by the existing descriptors. See also the SI (appendix A3) for the detailed procedures of the descriptor computations and more discussions.
Mx
O2 families with the same layered structure as LiCoO2 but partly substituted with different transition metals M. Since these partial substitutions are represented as supercells, the system's apparent size is larger than the original unit cells. Our approach is unaffected by these apparent differences and can recognise the essential similarities. While most of our list is filled with lithium oxides, the other two lists obtained by the existing descriptors do not suggest this consistent trend. These results suggest that our model recognises the concept of lithium-ion battery cathodes, which is not captured by the existing descriptors. See also the SI (appendix A3) for the detailed procedures of the descriptor computations and more discussions.
Next, we examined lithium-ion battery materials, which substantially support our lives of today. This technology has been developed through the discovery of new materials and the understanding of their structure-composition-property-performance relationships and is now bottlenecked by the cathodes (positive electrodes) in terms of the energy density and production cost [5]. We therefore studied the neighbourhoods of LiCoO2, the first yet most dominant cathode material [5]. Impressively, two of the three leading cathode material groups, namely, the layered, spinel families [5] (see figure 6(b) for visualisations), were identified in the neighbourhoods. Specifically, similar to LiCoO2, a family of layered LiMO2, with M being transition metals, were found within the top-10 neighbourhoods of LiCoO2 (see table 2), including important battery materials LiNiO2 families. Spinels as another important family were found as LiNi2O4 at the 51th neighbour and LiCo2O4 in the 200s neighbours. The polyanion family, the remaining one of three major cathode families, were not placed in the vicinity of LiCoO2 but formed a distinctive cluster at the top edge in figure 3(a). Interestingly, all of these materials were developed by the group of Nobel laureate John Goodenough [5]. This fact suggests that the embeddings capture conceptual similarity among the battery materials that previously required one of the brightest minds of the time to be discovered.
Note that our method properly links substituted materials and the original material without being confused by ad hoc supercell expression (e.g. Li4Co3NiO8 = LiCo0.75Ni0.25O2). This advantage is particularly noticeable in comparison with embeddings constructed using conventional features (table 2). This result indicates that our approach can recognise the essential structural features without being affected by superficial differences (i.e. the number of atoms or the size of the unit cell).
Additionally, we analysed the vicinities of magnetic materials, including 2D ferromagnets, which are attracting much attention for their interesting properties [41], and commercial samariumâcobalt (SmâCo) permanent magnets. Again, the embeddings capture meaningful similarity in these material classes, as shown in figures 7 and 8, which is often not evident to non-specialists (see appendix A1 in the supplementary information (SI) for more discussions and detailed results).
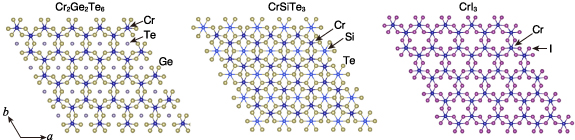
Figure 7. Crystal structures of the 2D ferromagnet Cr2Ge2Te6 and its neighbours in the embedding space. The double discoveries of 2D ferromagnets in 2017, after long questioning their existence, are gathering great interest from the magnetic materials community [41]. When we analysed the neighbourhoods of one of these 2D ferromagnets, Cr2Ge2Te6 (mp-541449), our embedding space successfully captured CrSiTe3 (mp-3779), a compound known as a potentially 2D-ferromagnetic insulator, as the first neighbour and even the other 2D ferromagnet CrI3 (mp-1213805) as the 15th neighbours among 122â543 materials. More detailed results and discussions are given in the SI (appendix A1).
Download figure:
Standard image High-resolution image
Figure 8. Crystal structures of the Sm2Co17 permanent magnet and its neighbours in the embedding space. Here we highlight two compounds in the neighbourhood list of Sm2Co17: SmCo5 and SmCo12. Sm2Co17 and SmCo5 are the two major components in SmâCo magnets often used in high-temperature environment, whereas SmCo12 is one of the compounds with the so-called 1â12 structure that has been drawing attention for its potential for permanent magnets. In the neighbourhoods of Sm2Co17 (mp-1200096) in our embedding space, we found SmCo12 (mp-1094061) as the 255th neighbour and SmCo5 (mp-1429) around the top 0.5% neighbourhoods. It is well known in the community that the crystal structures of Sm2Co17, SmCo5, and SmCo12 have close connections with each other [4]. However, without the literature context and proper visualisation, it is difficult for a human analyst to recognise these connections. More detailed results and discussions are given in the SI (appendix A1).
Download figure:
Standard image High-resolution imageThese in-depth analyses across diverse materials consistently support the conclusion that our ML model recognises similar functionalities of materials behind different structures without being explicitly taught to do so. We anticipate that when a material with beneficial properties is found, we may be able to screen for new promising candidates based on the conceptual similarities captured in this embedding space.
2.3. Performance validation as a materials descriptor
Here we provide quantitative insight into characteristics of embeddings. Particularly, we analyse the performance of predicting material properties using trained embeddings as input. As we are more interested in predicting functional material properties, we conducted a binary classification task of materials concepts, in which an ML model predicts whether a material belongs to a particular material class or not.
We expect that our embeddings contain the information of materials concepts. If so, we can rapidly screen materials with a desired concept from a material database by combining the embeddings with an ML model. However, properly labelling materials with their concepts requires experiments or consideration by experts, and thus the number of available labelled data for a given concept is likely to be limited. Therefore, as a benchmark and a use case for our embeddings, we evaluated the materials concept classification in the settings of few training data.
As benchmark materials, we used superconductors and thermoelectric materials for their complex and interesting properties. We used the Crystallography Open Database (COD) as the data source. The number of positive data used for training was 469 for superconductors and 286 for thermoelectric materials. Embeddings of these materials were obtained by the crystal structure encoder trained on the MP dataset via deep metric learning, and were used as input to a random forest classifier. As a baseline for comparison, we used latent feature vectors of crystal graph convolutional neural network (CGCNN) trained for total energy prediction, as in appendix C. We evaluated the prediction performance with leave-p-groups-out cross-validation while varying the training data size. Here, both the training and testing splits were made to contain balanced positive and negative samples.
As shown in figure 9, the classifier using our embeddings obtained good classification performance for both superconductors and thermoelectric materials. In particular, when the number of training data is very small (around 10), our method shows significantly better performance than the baseline. We will more discuss these results in the next section.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. The prediction performance of materials concepts. For superconductors and thermoelectric materials, embeddings obtained by our DML approach show higher performance especially when the size of the training dataset is very small. The embeddings of the baseline were latent vectors of CGCNN trained to predict total energy from crystal structures, as done by Xie et al [12].
Download figure:
Standard image High-resolution image{kind=link}
{kind=link}
3. Discussions
As assumed, materials concepts were exposed spontaneously in an abstract space. As we confirmed in the numerical evaluations of the training task of metric learning (appendix B in SI), this space was shown to successfully unify the two complementary factors of crystal structures. We hypothesise that these remarkable properties of our embeddings were made possible by the following two key features of our method that are distinctive from the existing material embedding methods [25â29]. First, we used deep metric learning, which directly optimises the spatial arrangements of the embedding vectors via a loss in terms of the Euclidean distances between them. This procedure is critically different from the existing methods [25â29], which learn embeddings indirectly as DNN's latent vectors. Although these latent vectors should encode essential information about materials, the explicit metric optimisation of embeddings is equally important for map creation and similarity learning. Second, our self-supervised learning is enabled by exploiting two forms of inputs expressing complementary structural characteristics: a set of atoms in the unit cell with their connections as the local characteristics and the XRD pattern, which is essentially a Fourier transformed crystal structure [1], as the periodic characteristics. Representation learning is known to be generally more well-informed when diverse multi-modal data are used for training [44]. In contrast to approaches that rely on single forms of materials data expression [25â29], our model benefits from learning across two forms of expression, or cross-modal learning.
The results of the materials concept classification (figure 9) clearly support these hypotheses. Remind that the baseline method (CGCNN [12]) learns embeddings as latent vectors in a DNN with only crystal structures as input, whereas our method uses the same DNN but trains it along with another DNN for XRD patterns in cross-modal deep metric learning. Thus, the performance advantage of our method directly indicates the benefit of the proposed cross-modal deep metric learning approach. We believe that our method using both crystal structures and XRD patterns helped the ML model to capture local motifs and lattice more effectively, which contributed to better learning of structural patterns correlated to material functionality and thus better recognition of materials concepts. We expect that incorporating more diverse structure representations of materials such as electronic structure into our multi-modal learning framework will further benefit the representation learning of materials. We leave such extensions as future work.
To provide more insight into the difference between our method and the baseline (CGCNN), we further analysed the performance of these methods for physical property prediction (see appendix C in SI for details). Similarly to the materials concept classification, we trained random forest models to predict materials properties, such as total energy, space group, and density, from learned embeddings. Our embedding outperformed the baseline in predicting density and space group and performed comparably in total energy and magnetisation (figure S1 in SI). This result confirms that our embeddings indeed capture lattice information in crystal structures more effectively than the single-modal baseline using only crystal structures. Performing comparably in total energy prediction is also notable, because the embeddings of the baseline are trained to specifically predict total energy itself using rich supervision from density functional theory calculations while our embeddings are not.
A major interest in the proposed method given its good predictive power is whether it has potential utility for new material discovery. To investigate such possibilities, we conducted a simple test to see if our model can re-discover superconductors known in the literature but not included in the training dataset. To this end, we borrowed the COD's superconductors from the concept classification (figure 9) and, after removing overlaps with the MP's training dataset, we mapped their embeddings in the MP's embedding distribution presented in figure 3. As shown in appendix E, these COD's superconductors are most intensively concentrated around the superconductor cluster in the MP's training materials, despite the fact that these COD's materials are novel to the model. This result suggests a screen method of new candidate materials by using our model trained on a database of known materials.
Another notable strength of our method over existing material embedding methods is that it does not require costly annotations and can be trained using only primitive structural information (i.e. crystal structures and their XRD patterns). This makes our method applicable to a wide range of datasets. Even when annotations are available, our self-supervised approach will benefit many users as a means of pre-training. Pre-training is a general ML technique performed on a large-scale dataset to help an ML model for other tasks where annotated training data are limited [45]. Our self-supervised learning is suitable for this purpose, because it can be performed given only crystal structure data and can thus utilise various material databases at scale.
When compared to classic material descriptors such as the Coulomb matrix variants [22], our method has advantages in terms of its scalability and ability to capture high-level material properties. See tables 1, 2 and the SI (appendix A3) for analysis results and more discussions.
Since the focus of our study was on learning material similarity from unannotated structural data, the resulting map requires manually interpreting clusters on the basis of our knowledge of materials concepts. Interestingly, a word2vec model [26] has been applied to text symbols appearing in the materials science literature, thus learning relationships such as the connections between 'Fe' and 'metal' and between 'SmâCo' and 'magnet'. Use of this technique may further automate the interpretation of our results with literal knowledge.
4. Conclusions and broader impacts
In summary, we have demonstrated the self-supervised learning of material embeddings solely from crystal structures using DNNs. Careful inspection of the embedding space, in terms of both the global distribution and local neighbourhoods, has confirmed that the space recognises functionality-level material similarity or materials concepts. Our techniques for the materials space visualisation and the similarity evaluation between crystal structures will be useful for discovering new underlying relationships among materials and screening for new promising material candidates. Since these techniques are not strongly affected by human bias, they could give rise to a new view of materials that can stimulate efforts to break through our knowledge barriers.
Our result is also applicable to material retrieval systems that can search for conceptually similar materials in a database given a query material. This approach will enable us to rediscover materials that have never been recognised to have preferable properties.
Furthermore, constructing a functionality-aware representation space of crystal structures is a first step towards the inverse design of materials [8, 46], a grand challenge of materials informatics. This workflow would allow us to design materials in the functionality space and inversely map the functionality attributes to synthesisable crystal structures with the desired properties. We hope that this study will pave the way for breakthroughs in the ML-assisted discovery and design of materials.
5. Methods
5.1. Data acquisition and pre-processing
We used the Materials Project as the data source for this study. We collected data for up to quintet systems, excluding monatomic crystals, on 8 July 2020, using the Material Project APIs, which resulted in a total of 122â543 materials (93% of the source collection) as our targets. We additionally queried thermodynamic stability material attributes on 14 October 2020. We used VESTA [47] for crystal structure visualisation. We calculated the XRD patterns using pymatgen [48]. The x-ray wavelength was set to 1.54â184âÃ
(Cu K ), and the 2θ angle ranged from 10â to 110â with a step size of 0.02â; thus, 5000-dimensional vectors of 1D-structured XRD patterns were produced. To ease the learning process, the intensity scale of each XRD pattern was normalised by setting the maximum intensity to 1.
), and the 2θ angle ranged from 10â to 110â with a step size of 0.02â; thus, 5000-dimensional vectors of 1D-structured XRD patterns were produced. To ease the learning process, the intensity scale of each XRD pattern was normalised by setting the maximum intensity to 1.
5.2. Neural network architecture
As illustrated in figure 2(a), we used two types of DNNs as embedding encoders. For the crystal-structure encoding, we need to convert a set of arbitrary number of atoms (i.e. the atoms in the unit cell) into a fixed-size embedding vector in a fashion invariant to the permutation of atom indices. For this purpose, we used CGCNNs [12]. As input to CGCNN, the 3D point cloud of the atoms in the unit cell is transformed into a graph of atoms whose edge connections are defined by their neighbours within a radius of 8âà . The atoms in the graph are represented as atom feature vectors and are transformed into a single fixed-size feature vector via three graph convolution layers and a global pooling layer. For the XRD patterns, we used a standard feed-forward 1D convolutional neural network designed following existing studies on XRD pattern encoding [49]. At the end of each network, we used three fully connected layers to output 1024-dimensional embedding vectors. Since one of these encoders is supervised by the output of the other in our self-supervised learning approach, training them simultaneously tends to be unstable compared to standard supervised learning. To stabilise the training process, we found that batch normalisation [50] is essential after every convolutional/linear layer in both networks except for the final linear output layers. We discuss this further in the SI (appendix B). Further details of our network architecture are provided in the SI (tables S6 and S7 in appendix D) and our ML model codes.
5.3. Training procedures
In each training iteration, we processed a batch of N input material samples. Let
x
i
and
y
i
be a pair of embedding vectors produced for the ith crystal structure in a batch and its XRD pattern, respectively. For each positive pair  , we randomly drew two kinds of negative samples
, we randomly drew two kinds of negative samples  and
and  , representing a crystal structure and an XRD pattern, respectively, from the batch to form two triplet losses:
, representing a crystal structure and an XRD pattern, respectively, from the batch to form two triplet losses:


where the negative sample  was chosen from
was chosen from  to produce a positive-valued loss,
to produce a positive-valued loss,  , and
, and  was chosen similarly from
was chosen similarly from  (see also figure 2(b) for illustrations). Here, mâ>â0 is a hyperparameter called the margin. Equation (1) essentially requires that for each embedding
y
i
, its negative samples
(see also figure 2(b) for illustrations). Here, mâ>â0 is a hyperparameter called the margin. Equation (1) essentially requires that for each embedding
y
i
, its negative samples  are cleared out of the area surrounding
y
i
having the radius of the positive-pair distance
are cleared out of the area surrounding
y
i
having the radius of the positive-pair distance  (red circle in the top-right part of figure 2(b) plus the margin m (yellow area in the figure). Equation (2) is defined similarly. These losses are thus to ensure, given an embedding as a query, that its paired embedding is retrievable as the query's nearest neighbour. Note that the choice of the margin m is quite flexible because its value is relevant only to the scales of the embeddings, which are unnormalised and arbitrarily learnable. Here, m = 1. Our bidirectional triplet loss was then computed as the average of the losses for all samples in the batch as follows:
(red circle in the top-right part of figure 2(b) plus the margin m (yellow area in the figure). Equation (2) is defined similarly. These losses are thus to ensure, given an embedding as a query, that its paired embedding is retrievable as the query's nearest neighbour. Note that the choice of the margin m is quite flexible because its value is relevant only to the scales of the embeddings, which are unnormalised and arbitrarily learnable. Here, m = 1. Our bidirectional triplet loss was then computed as the average of the losses for all samples in the batch as follows:

This expression is similar to but simpler in form than a loss expression previously used in cross-modal retrieval [51].
We optimised the loss function using stochastic gradient descent with a batch size N equal to 512. Using the Adam optimiser [52] with a constant learning rate of 10â3, we conducted iterative training for a total of 1000 epochs for all target materials in the dataset. The training took approximately one day using a single NVIDIA V100 GPU. For details regarding our strategies for validating the trained models and tuning the hyperparameters (e.g. choices of the embedding dimensionality and training batch-size), see appendix B and table S5 in the SI.
5.4. Data acquisition for the concept classification tasks
For the materials concept classification, we collected the crystal structure data of superconductors and thermoelectric materials from COD. To collect positive samples for each category, we retrieved material entries containing certain keywords in their paper titles as positive samples. Specifically, the entries including 'superconductor' or 'superconductivity' in their titles were regarded as superconductors, and the entries including 'thermoelectric' or 'thermoelectricity' were regarded as thermoelectric materials. The same number of material entries without these keywords were randomly collected and used as negative samples.
Acknowledgment
This work is partly supported by JST-Mirai Program, Grant Nos. JPMJMI21G2 and JPMJMI19G1. Y S is supported by JST ACT-I Grant Number JPMJPR18UE.
Data availability statement
The materials data retrieved from the Materials Project, the trained embeddings of these materials, and the trained ML model weights are available at the figshare repository [53]. The list of the target materials used in this study, the lists of the neighbourhood search results, and interactive web pages for exploring the materials map visualisation and analysing local neighbourhoods are available in the GitHub repository (https://github.com/quantumbeam/materials-concept-learning).
Author contributions
Y S, Y U, and K O conceived the idea for the present work. Y S and T T carried out the numerical experiments. All authors discussed the results and wrote the manuscript together.
Competing interests
The authors declare no conflicts of interest associated with this manuscri.
Supplementary data (1.0 MB PDF)