

Highlights
-
•
We investigated the ACE2 levels after infection with influenza A (H1N1) virus.
-
•
Influenza infection results in downregulation of ACE2 protein levels that was dispensable for viral replication.
-
•
ACE2 downregulation was most likely related to ACE2 protein degradation by proteasome pathway rather than ACE2 shedding.
-
•
The neuraminidase of influenza virion results in ACE2 cleavage.
Keywords: Influenza, ACE2, Downregulation, Neuraminidase
Abstract
Influenza A (H1N1) virus, a high-risk infectious pathogen, can cause severe acute lung injury leading to significant morbidity and mortality. Angiotensin-converting enzyme 2 (ACE2), a negative regulator of the renin-angiotensin system (RAS), plays a protective role in pathogenesis of acute lung injury. Here, we showed that ACE2 protein levels were significantly downregulated after infection with H1N1 viruses but was dispensable for viral replication. ACE2 protein downregulation was most likely related to ACE2 protein degradation by proteasome pathway rather than ACE2 shedding. Finally, we found that ACE2 cleavage could be regulated by influenza neuraminidase (NA), which was fundamentally different from the classically sheddase-induced proteolytic cleavage of ACE2.
1. Introduction
Influenza A (H1N1) virus infections can cause severe acute lung injury leading to significant morbidity and mortality (Carmona et al., 2011, Glezen, 1982, Kasowski et al., 2011). The renin-angiotensin system (RAS) plays a key role in pathogenesis of acute lung injury (Jerng et al., 2007, Kuba et al., 2006, Marshall et al., 2004). Angiotensin-converting enzyme 2 (ACE2), a negative regulator of the RAS, protects against acute lung injury in several animal models of acute respiratory distress syndrome (ARDS) (He et al., 2007, Imai et al., 2005, Li et al., 2008b). ACE2 was identified as a functional receptor for the SARS coronavirus (SARS-CoV), and mediated efficient virus replication (Hamming et al., 2004, Li et al., 2004, Li et al., 2003, Li et al., 2005). ACE2 expression was markedly downregulated in acute lung injury of mice caused by infection with SARS-CoV. The virus replication was effective in ACE2-transfected 293T cells, but could be blocked by anti-ACE2 antibodies on Vero E6 cells (Li et al., 2003). However, little is known about the mechanism of ACE2 in pathogenesis of influenza-induced acute lung injury. If ACE2 expression levels were altered after influenza virus infection has not been confirmed yet.
In this study, we investigated ACE2 expression levels in vitro after infection with influenza A (H1N1) virus including PR8, CA07, H5N1 and H3N2 strains, and found that ACE2 protein levels were significantly downregulated on virus infection in either CNE-2E or 293T-ACE2 stable cell lines. ACE2 protein downregulation was most likely related to ACE2 protein degradation by proteasome pathway rather than ACE2 shedding. Further, we put forward a mechanism of ACE2 cleavage by influenza neuraminidase (NA) different from proteolytic cleavage of ACE2 by sheddase.
2. Materials and methods
2.1. Viruses and cells
The influenza viruses used in this study were influenza A (H1N1) virus strains: A/Puerto Rico/8/1934 (PR8), A/California/07/2009 (CA07), A/Anhui/01/2005 (H5N1) and A/victoria/201/2009 (H3N2). The strains were inoculated in 9–11-day-old SPF chicken embryos. The median tissue culture infective dose (TCID50) was determined by using MDCK cells according to the Reed–Muench method (Reed and Muench, 1938). All manipulations with live virus were performed by the standard operating procedures of the approved biosafety level-3 animal facilities. The human nasopharyngeal carcinoma cell line CNE-2Z and human embryonic kidney cell line 293T were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin-streptomycin at 37 °C with 5% CO2.
2.2. Treatment protocols
Cells were infected with influenza virus at the indicated multiplicity of infection (MOI) at 37 °C with 5% CO2 for 1 h. After this time, cells were washed twice with PBS and incubated with fresh DMEM containing 10% FBS, 100 units/ml penicillin-streptomycin at 37 °C with 5% CO2 for the indicated times. Then, cell lysates or supernatants were obtained for western blotting assays. The titer of virus was determined by standard plaque assay (Tobita et al., 1975).
2.3. Western blotting assay
Western blotting assay was performed as previously described (Yang et al., 2011). Protein extractions from cell lysates were obtained with RIPA buffer mixed protease inhibitor cocktail (Thermo) according to the manufacturer's instructions. Protein extractions were boiled in SDS-PAGE loading buffer for 5 min, and resolved on a SDS-PAGE, then transferred onto PVDF membrane. Membranes were blocked with 2% albumin from chicken egg white (Sigma) for 1 h, and incubated with anti-ACE2 antibodies (R&D) and anti-β-actin antibodies (Sigma) overnight at 4 °C. After triple wash with TBST, they were incubated with anti-goat horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz) for 1 h at room temperature. Final detection of protein was performed using the Signal Boost Immunoreaction Enhancer Kit (Merck Millipore). Protein levels were quantified by using Quantity One software (Bio-Rad).
2.4. Transfection with pcDNA3.1 plasmids
A full-length cDNA encoding human ACE2 protein was cloned into the pcDNA3.1-MycHisA vector. 293T cells were seeded in 24-well plate at 37 °C with 5% CO2 for 24 h. When 50% confluent, 293T cells were transfected with either pcDNA3.1-ACE2 or empty vector as control using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's instructions. After 48 h transfection, 293T cell line stably expressing ACE2 protein (293T-ACE2) were selected using 0.5 mg/ml G418 for two weeks.
For activities assays of influenza A (H1N1) virus NA (N1), Flag-tagged N1 gene of PR8 was cloned into the pcDNA3.1 vector to generate pcDNA-Flag-N1 plasmid, and Flag-tagged EGFP gene was inserted into the vector to generate pcDNA-Flag-EGFP as control. 293T-ACE2 cells were seeded in 24-well plate at 37 °C with 5% CO2. Once a 50% confluent, 293T-ACE2 cells were transfected with either pcDNA-N1 or pcDNA-EGFP. After 12 h transfection, N1 and EGFP positive expressions in 293T-ACE2 cells were identified by Western blotting with anti-Flag antibodies.
2.5. Flow cytometric assay
CNE-2Z cells infected with PR8 at 24 h post infection (pi) were collected and incubated with anti-ACE2 antibody for 2 h at 37 °C. After triple wash with PBS, they were incubated with Alexa Fluor 488-labeled secondary antibodies (Invitrogen) at room temperature for 1 h. Stained cells were rinsed twice with PBS, acquired by a FACScan cytometer and analyzed with FACSDiva software (BD).
2.6. RNA isolation and quantitative real-time PCR
Quantitative real-time PCR was performed as previously described (Yang et al., 2011). Total RNA was extracted from cultured cells with TRIzol reagent (Invitrogen) according to the manufacturer's instructions (Chen et al., 2002). 5 μg of total RNA was treated with RNase free DNase I (Promega) for 30 min at 37 °C. Complementary DNA was generated by reverse transcription with PrimeScript RT Master Mix (TaKaRa) according to the manufacturer's directions. 10 μl of LightCycler 480 SYBR Green I Master (Roche) were mixed with template and primers. The real-time PCR for ACE2, GAPDH expression levels measurement were performed in triplicate wells of a 96-well reaction plate on a LightCycler 480 system (Roche). The primer sequences of ACE2 were 5′-CATTGGAGCAAGTGTTG-GATCTT-3′ (sense) and 5′-GAGCTAATGCATGCCATTCTCA-3′ (antisense) (Li et al., 2008a); TACE primer sequence, 5′-CAGCACAGCTGCCAAGTCATT-3′ (sense) and 5′-CCAGCATCTGCTAAGTCACTTCC-3′ (antisense); GAPDH primer sequence, 5′-CCCCTGGCCAAGGTCATCCATGAC-3′ (sense), and 5′-CATACCAGGAAATGAGCTTGACAAAG-3′ (antisense). The quantification data were analyzed with LightCycler Software v.1.5 (Roche).
2.7. Small hairpin RNA (shRNA) expression
For shRNA-mediated knockdown, double-stranded oligonucleotides targeting TACE (shTACE) or GFP (shCtrl) were inserted into the retroviral vector pSIREN-RetroQ (kindly provided by Professor Depei Liu). CNE-2Z cells were infected with retrovirus expressing shTACE or shCtrl for 48 h, and then TACE mRNA expression levels were analyzed by real-time PCR. CNE-2Z cells were infected with PR8 or CA07 virus after 48 h retroviral infection. The cell lysates were obtained at 24 hpi, and then ACE2 protein levels were detected by Western blotting.
2.8. Transfection with small interfering RNA (siRNA) targeting ACE2
For ACE2 knockdown experiment, two different siRNAs against different regions of the target gene ACE2 were synthetized. Sequences were as follows: siACE2#1, 5′-GGCCAUUAUAUGAAGAGUA-3′, #2, 5′-CCAUCUACAGUACUGGAAA-3′. CNE-2Z cells were transfected with 20 nM ACE2 siRNAs using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions. Forty-eight hours after transfection, ACE2 expression levels were analyzed by Western blotting with anti-ACE2 antibodies. After 48 h transfection, CNE-2Z cells were infected with or without PR8 virus, and then influenza nuclear protein (NP) expression levels at 12 hpi were detected by Western blotting with anti-NP antibodies (Upstate).
2.9. Statistical analyses
Statistical analysis of the results was performed in Excel and GraphPad Prism software (version 4.0, GraphPad). Data was shown as means ± S.E.M. Statistical significance in comparing two means was tested with the unpaired Student's t-test. Statistical significances are indicated as *P < 0.05.
3. Results
3.1. Influenza A (H1N1) virus infection results in a decline in ACE2 protein levels
We investigated ACE2 protein levels after influenza A (H1N1) virus infection in CNE-2Z cells. The cells were infected with PR8 or CA07 at the multiplicity of infection (moi) of 1 and 5. Control cells were mock-infected with allantoic fluid from SPF chick embryos. The cell lysates were obtained at 12 hpi and 24 hpi, and then ACE2 protein levels were detected by Western blotting. The results showed that ACE2 was decreased after infection with two strains in a dose-dependent manner (Fig. 1a and b). ACE2 protein levels showed clear decline at a high moi of 5 at 24 hpi. The decline of ACE2 protein levels was also observed in 293T-ACE2 infected with either PR8 or CA07 viruses (Fig. 1d). Further, ACE2 protein levels in CNE-2Z cells were examined by flow cytometry. The fluorescence intensity of ACE2 was decreased after PR8 infection at 24hpi (Fig. 1c). Then, the ACE2 mRNA levels in CNE-2Z cells were analyzed by real-time PCR; However, the ACE2 mRNA level was not altered after PR8 infection compared with mock-infected cells (Fig. 1e), and as a control, there was hardly any ACE2 mRNA expression in A549 cell lines. These results suggested that influenza virus infection downregulated ACE2 protein levels.
Fig. 1.
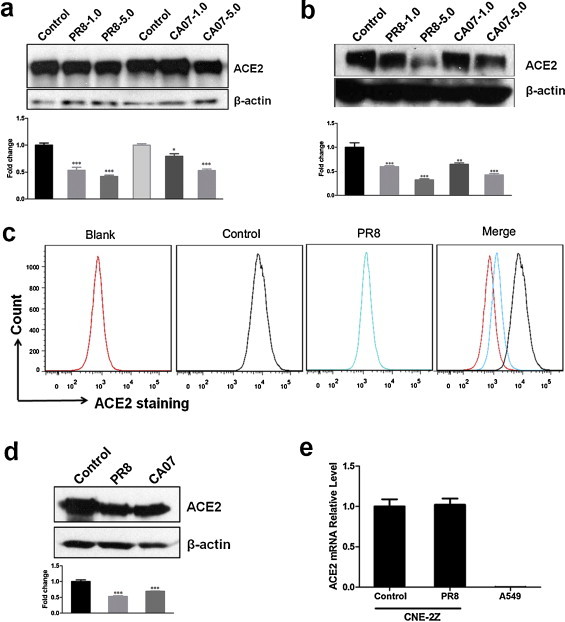
Influenza A (H1N1) virus infection results in a decline of ACE2 protein levels in cell lysates of CNE-2Z and 293T cell line stably expressing ACE2. The levels of ACE2 protein were detected by Western blotting. Densitometric analysis relative to β-actin levels was expressed as fold change. (a, b) The cell lysates of CNE-2Z were obtained at 12 hpi (a) and 24 hpi (b) with PR8 or CA07 viruses at moi of 1 and 5. Control cells were mock-infected with AF. (c) Flow cytometric analysis of CNE-2Z cells infected with PR8 or AF (control) at 24 hpi using anti-ACE2 antibody. (d) The cell lysates of 293T-ACE2 were obtained at 24 hpi with PR8 or CA07 viruses at moi of 5. (e) Total RNA of CNE-2Z after 24 hpi with PR8 or AF (control) was obtained. PR8-infected A549 cells were used as cell control. ACE2 mRNA level was analyzed by real-time PCR. All graphs represent the means ± S.E.M. *P < 0.05 were considered statistical significant.
3.2. Influenza virus-induced ACE2 protein downregulation was not due to ACE2 shedding, and it was not dependent on TACE
We next asked if ACE2 protein decline was due to ACE2 shedding into cell culture supernatants. Firstly, the cell supernatants of CNE-2E and 293T-ACE2 were obtained at 24hpi after infection with PR8 or CA07 strains at a moi of 5; Control cells were mock-infected with AF. It was showed that ACE2 shedding into supernatants both from mock-infected cells and virus-infected cells (Fig. 2a and b). The ACE2 protein levels in cell supernatants were actually decreased rather than increased after infection with PR8 or CA07, which suggested that ACE2 shedding was not responsible for influenza virus-induced ACE2 protein decline.
Fig. 2.
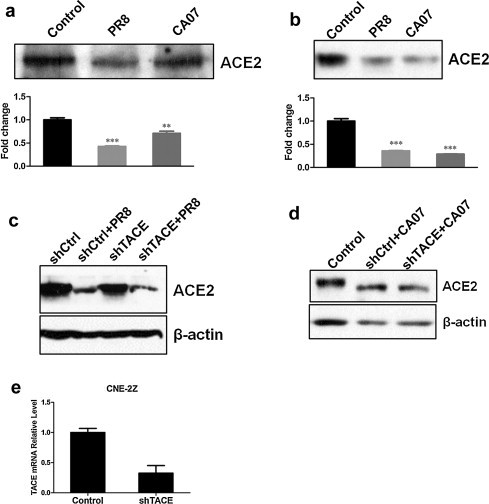
ACE2 shedding was not responsible for influenza virus-induced ACE2 protein downregulation, and TACE knockdown did not affect the levels of ACE2 downregulation. The cell culture supernatants of CNE-2E (a) and 293T-ACE2 (b) cells were obtained at 24 hpi after infection with PR8 or CA07 strains at a moi of 5; Control cells were mock-infected with AF. The levels of ACE2 protein were detected by Western blotting. Densitometric analysis normalized to the corresponding control levels was expressed as fold change. (c, d) CNE-2Z cells were infected with the retroviruses either expressing shTACE or shCtrl. After 48 h, cells were infected with PR8 or CA07. The cell lysates were taken at 24 hpi, and then ACE2 protein levels were detected by Western blotting. (e) Real-time PCR analyzed TACE mRNA expression levels after a 48 h of retroviral transduction. All graphs represent the means ± S.E.M. *P < 0.05 were considered statistical significant.
Previous study has demonstrated that binding of SARS-S to ACE2 induced ACE2 shedding in a tumor necrosis factor alpha (TNF-α)-converting enzyme (TACE)-dependent manner (Haga et al., 2008). So, we further tested if ACE2 protein levels in cell lysates would be increased after TACE knockdown. In order to efficiently knockdown TACE expression, CNE-2Z cells were infected with retrovirus expressing shTACE. TACE mRNA expression levels were analyzed by real time-PCR after 48 h retroviral transduction; and presented up to 70% knockdown (Fig. 2e). However, ACE2 protein levels were not altered in cells infected with retrovirus expression shTACE compared with those expression shCtrl (Fig. 2c and d), which suggested that ACE2 downregulation induced by influenza infection did not depend on TACE.
3.3. ACE2 protein decline in cell lysates was related to ACE2 protein degradation by proteasome pathway
ACE2 Shedding could not provide convincing explanation for the previously observed ACE2 protein decline in cell lysates. Then, we wonder whether ACE2 protein decline was due to ACE2 degradation by proteasome pathway. So, we treated CNE-2Z cells with MG132, a 26S proteasome inhibitor (Lee and Goldberg, 1998), or solvent DMSO as control. The results showed that ACE2 protein levels increased after MG132 treatment (Fig. 3a). Moreover, MG132-pretreated CNE-2Z cells accumulated much more ACE2 protein after PR8 infection compared with DMSO-pretreated control cells (Fig. 3b), which suggested that ACE2 protein decline induced by influenza virus was related to protein degradation. Accordingly, ACE2 protein degradation by proteasome pathway most likely accounted for the ACE2 protein decline after H1N1 infection as previously observed.
Fig. 3.
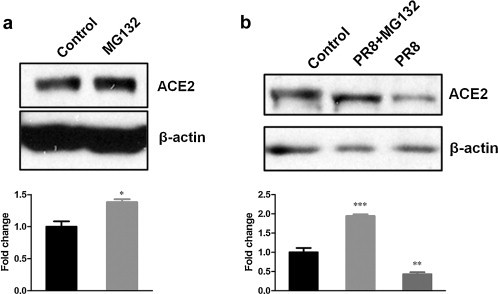
ACE2 protein decline in cell lysates was related to ACE2 protein degradation by proteasome pathway. (a) ACE2 protein levels in CNE-2Z cells treated with 5 μM MG132 or solvent DMSO as control for 8 h. (b) CNE-2Z cells were pretreated with MG132 (lane 2) or solvent DMSO (lane 1, 3) for 2 h, and then infected with PR8 virus (lane 2, 3) or AF (lane 1). The levels of ACE2 protein were detected by Western blotting. Densitometric analysis relative to β-actin levels was shown as fold change. All graphs represent the means ± S.E.M. *P < 0.05 were considered statistical significant.
3.4. ACE2 downregulation did not affect virus replication.
It is still not clear whether or not the decline of ACE2 have an effect on virus replication. CNE-2Z cells were transiently transfected with two different siRNAs against different regions of the target gene ACE2: siACE2#1 and siACE2#2. As shown in Fig. 4a, the application of siACE2#1 and siACE2#2 did practically knock down the ACE2 gene expression. After 48 h of siRNA transfection, CNE-2Z cells were infected with PR8 virus. However, influenza NP protein levels were not altered significantly after 12 h infection (Fig. 4b). Further, the release of viral particles was examined by plaque assay (Fig. 4c), and there was little difference in progeny virus titer between groups. The results suggested that the ACE2 downregulation might not have an effect on virus replication.
Fig. 4.
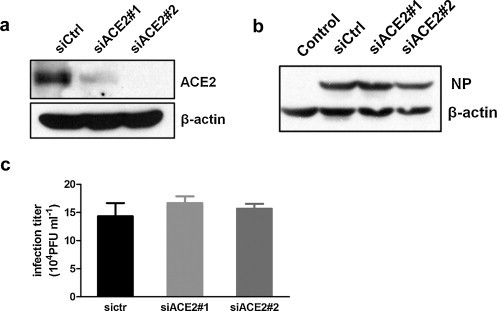
ACE2 downregulation did not affect virus replication. (a) CNE-2Z cells were transiently transfected with negative control siRNA (siCtrl) or two ACE2-specific siRNAs (siACE2#1 and siACE2#2). After siRNA transfection for 48 h, the cells lysates were analyzed by Western blotting with anti-ACE2 antibodies. (b) CNE-2Z cells were infected with or without PR8 virus after 48 h siRNA transfection. Influenza NP expression levels were detected by Western blotting after 12 h infection. (c) CNE-2Z cells were infected with PR8 after 48 h siRNA transfection, and then progeny virus titers were determined by plaque assays. All graphs represent the means ± S.E.M.
3.5. ACE2 protein molecular weight declined after influenza virus infection
We demonstrated that ACE2 protein downregulation was related to ACE2 protein degradation by proteasome pathway, but the mechanisms of ACE2 protein downregulation are still uncompletely clarified. ACE2 protein molecular weight seemed a slight decrease after influenza virus infection by a shift in electrophoretic mobility (Fig. 1, Fig. 3), which aroused us to further confirm whether ACE2 protein molecular weight was declined by influenza infection and when it occurred. The electrophoresis time was increased. The results showed that ACE2 protein molecular weight was decreased after PR8 and CA07 strains infection (Fig. 5a and b). Further, ACE2 protein size at indicated time points was examined. The results suggested that the decrease of ACE2 protein molecular weight occurred very soon after infection, followed by ACE2 protein degradation (Fig. 5c). But the size of reduced protein was not decreased anymore after subsequent infection. In addition, a similarly effect of ACE2 shift was observed by infection with other influenza subtypes, including H5N1 and H3N2 (Fig. 5d).
Fig. 5.
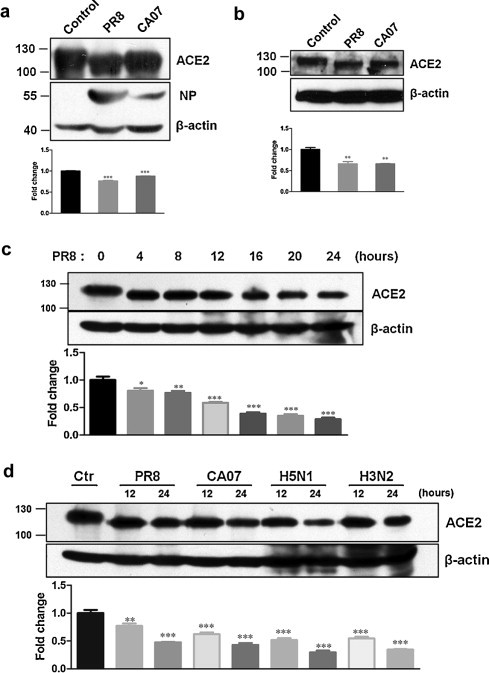
ACE2 protein molecular weight was declined after virus infection. The cell lysates of CNE-2Z (a) and 293T-ACE2 (b) were obtained at 12 hpi after infection with PR8 or CA07 at a moi of 5. Control cells were mock-infected with AF. Influenza NP (a) was also examined. (c) The cell lysates of CNE-2Z were obtained at indicated time points after infection with PR8 at moi of 5. (d) CNE-2Z cells were infected with PR8, CA07, H5N1 and H3N2. The cell lysates were obtained at 12 hpi and 24 hpi. Control cells were mock-infected with AF. ACE2 protein was detected by Western blotting. Densitometric analysis normalized to β-actin levels was shown as fold change. All graphs represent the means ± S.E.M. *P < 0.05 were considered statistical significant.
3.6. Influenza A (H1N1) virus neuraminidase (N1) resulted in ACE2 molecular weight decline
According to above results that the decline of ACE2 protein size occurred very early after infection, we wondered whether there was a similar but extracellular effect on ACE2 molecular by treatment with influenza virions. Thus, recombinant ACE2 proteins of equal quality were directly coincubated with influenza virions or AF (control). The results showed that ACE2 protein levels were decreased after coincubation recombinant ACE2 proteins with PR8 or CA07 (Fig. 6a); and ACE2 protein molecular weight seemed to be declined. The results indicated that the shift of ACE2 molecular weight was most likely due to directly cleavage by influenza virions rather than cell surface proteases.
Fig. 6.
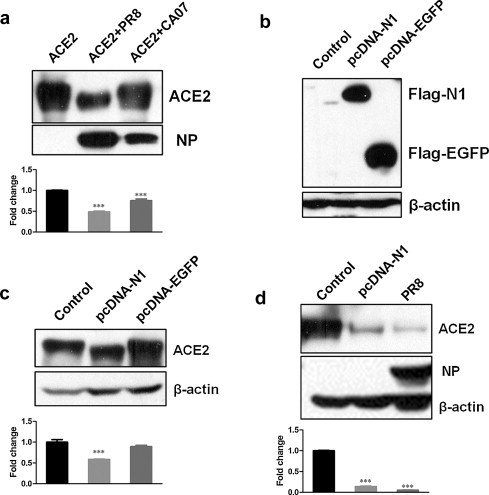
Influenza A (H1N1) virus neuraminidase (N1) resulted in ACE2 molecular weight decline. (a) Recombinant ACE2 proteins were coincubated with AF (lane 1) or PR8 (lane 2) and CA07 (lane 3) strains. After a 12 h coincubation, the levels of ACE2 protein and influenza NP were analyzed by Western blotting. Densitometric analysis normalized to ACE2 levels (lane 1) was shown as fold change. (b) N1 and EGFP positive expressions in 293T-ACE2 cells transfected with pcDNA-N1 or pcDNA-EGFP were verified by Western blotting with anti-Flag antibodies. Cells transfected with empty plasmid were used as control. (c) 293T-ACE2 cells were transfected with either pcDNA-N1 or pcDNA-EGFP. Untransfected parental 293T-ACE2 cells were used as negative control. After 48 h transfection, ACE2 protein levels were detected by Western blotting. Densitometric analysis normalized to β-actin was shown as fold change. (d) 293T-ACE2 cells were either transfected with pcDNA-N1 or infected with PR8 or AF (control). After 48 h, Western blotting analyzed the levels of ACE2 and NP. Densitometric analysis relative to β-actin was shown as fold change. All graphs represent the means ± S.E.M. *P < 0.05 were considered statistical significant.
Neuraminidase (NA), a major surface glycoprotein of the influenza virus, possesses critical enzymatic activity (Mitnaul et al., 1996). Hence, it is reasonable to suspect that ACE2 molecular weight decline might be related to NA enzyme activities. 293T-ACE2 cells were transfected with either pcDNA-N1 or pcDNA-EGFP to generate positive expression of N1 and EGFP (Fig. 6b). The results showed that cells transfected with pcDNA-N1 resulted in ACE2 molecular weight decline compared with those transfected either with or without pcDNA-EGFP plasmid (Fig. 6c). The downregulation of ACE2 protein levels could also be induced by pcDNA-N1 transfection into 293T-ACE2 cells compared with cells mock-infected with AF (Fig. 6d), which suggested that ACE2 molecular weight decline might due to NA enzymatic activity.
4. Discussion
In the present study we confirmed that ACE2 protein levels were downregulated on Influenza virus infection including PR8, CA07, H5N1 and H3N2 strains. SARS-CoV downregulated its receptor ACE2 and induced TACE-dependent ACE2 shedding. ACE2 shedding most likely accounted for SARS-CoV induced ACE2 downregulation (Glowacka et al., 2010). However, influenza virus-induced ACE2 decline was most likely related to ACE2 protein degradation by proteasome pathway rather than ACE2 shedding. Sheddases and some stimuli like virus infection regulate the release of ACE2 from the surface of cells (Jia et al., 2009). ACE2 shedding was actually not increased from influenza virus-infected CNE-2Z cells. Knockdown of TACE expression by shRNA did not affect ACE2 downregulation in this study. ACE2 as the receptor for SARS-CoV mediated efficient virus replication; ACE2 siRNA significantly decreased the SARS-CoV mRNA copy number (Haga et al., 2008). However, ACE2 knockdown did not attenuate influenza A virus replication in this study.
The ectodomain of a number of transmembrane proteins can be released as a soluble fragment by the action of cell surface proteases (Ehlers and Riordan, 1991, Hooper et al., 1997). Sheddase can generate proteolytic cleavage of membrane-associated ACE2 for its shedding (Jia et al., 2009, Lambert et al., 2005). In this study, it was identified that overexpression of Influenza A (H1N1) virus NA protein could reduce ACE2 protein molecular weights in 293T-ACE2 cells, but it did not increased ACE2 shedding (data not shown). Coincubation recombinant ACE2 proteins with PR8 or CA07 virus could also decrease both total ACE2 protein levels and molecular weights in vitro. These findings indicated that influenza A (H1N1) virus-induced ACE2 molecular weight decline might be the result of cleavage by enzyme activities of N1, which was different from ACE2 shedding. But it is still unknown whether the cleaved ACE2 by N1 retains its enzymatic activity.
Our results demonstrated that ACE2 protein can be cleaved directly by influenza virus NA protein, followed by degradation through proteasome pathway. But whether NA protein could cleave other proteins is still not clear. So, we cannot confirm the specificity of ACE2 protein cleavage by NA. Whether or not ACE2 levels were regulated by other proteins of influenza virions has not been confirmed yet. NA is a glycosidase that specifically promotes the cleavage of sialic acid from glycoprotein saccharide chains (Suttajit and Winzler, 1971). Whether NA catalyzed the hydrolysis of glycosidic linkages of ACE2 proteins are yet to be identified.
ACE2 has an important role in efficiently hydrolyzing the potent vasoconstrictor angiotensin II to angiotensin 1–7 in the body (Keidar et al., 2007). It has been reported that ACE2 was implicated in hypertension, cardiac function and diabetes (Clarke and Turner, 2012, Hamming et al., 2007). In mouse models of lung failure induced by acid aspriration or sepsis, ACE2 deficiency worsens acute lung injury (Imai et al., 2005). However, the regulation of ACE2 protein in influenza-induced lung injury has not been reported yet. In our study, ACE2 proteins can be reduced by influenza virions in vitro, and also by transient expression of NA proteins in cells. But whether a similar effect of ACE2 downregulation could be detected in influenza virus infected mouse models has not been confirmed yet. We think that it is very essential to figure out the meaning of ACE2 cleavage in pathogenesis and the effect of ACE2 deficiency in pathogenesis of influenza-induced acute lung injury in the future.
Acknowledgements
We are grateful to Depei Liu’ Lab of Peking Union Medical College for kindly providing the retroviral vector pSIREN-RetroQ.
References
- Carmona F., Carlotti A.P., Ramalho L.N., Costa R.S., Ramalho F.S. Evidence of renal infection in Fatal Cases of 2009 Pandemic Influenza A (H1N1) Am. J. Clin. Pathol. 2011;136(3):416–423. doi: 10.1309/AJCP1Y6LLHWSKYHW. [DOI] [PubMed] [Google Scholar]
- Chen G., Gharib T.G., Huang C.C., Taylor J.M., Misek D.E., Kardia S.L., Giordano T.J., Iannettoni M.D., Orringer M.B., Hanash S.M., Beer D.G. Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell. Proteomics. 2002;1(4):304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- Clarke N.E., Turner A.J. Angiotensin-converting enzyme 2: the first decade. Int. J. Hypertens. 2012;2012:307315. doi: 10.1155/2012/307315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers M.R., Riordan J.F. Membrane proteins with soluble counterparts: role of proteolysis in the release of transmembrane proteins. Biochemistry. 1991;30(42):10065–10074. doi: 10.1021/bi00106a001. [DOI] [PubMed] [Google Scholar]
- Glezen W.P. Serious morbidity and mortality associated with influenza epidemics. Epidemiol. Rev. 1982;4:25–44. doi: 10.1093/oxfordjournals.epirev.a036250. [DOI] [PubMed] [Google Scholar]
- Glowacka I., Bertram S., Herzog P., Pfefferle S., Steffen I., Muench M.O., Simmons G., Hofmann H., Kuri T., Weber F., Eichler J., Drosten C., Pohlmann S. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010;84(2):1198–1205. doi: 10.1128/JVI.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga S., Yamamoto N., Nakai-Murakami C., Osawa Y., Tokunaga K., Sata T., Sasazuki T., Ishizaka Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. U. S. A. 2008;105(22):7809–7814. doi: 10.1073/pnas.0711241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming I., Cooper M.E., Haagmans B.L., Hooper N.M., Korstanje R., Osterhaus A.D., Timens W., Turner A.J., Navis G., van Goor H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007;212(1):1–11. doi: 10.1002/path.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamming I., Timens W., Bulthuis M.L., Lely A.T., Navis G., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203(2):631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Han B., Mura M., Xia S., Wang S., Ma T., Liu M., Liu Z. Angiotensin-converting enzyme inhibitor captopril prevents oleic acid-induced severe acute lung injury in rats. Shock. 2007;28(1):106–111. doi: 10.1097/SHK.0b013e3180310f3a. [DOI] [PubMed] [Google Scholar]
- Hooper N.M., Karran E.H., Turner A.J. Membrane protein secretases. Biochem. J. 1997;321(Pt 2):265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., Kuba K., Rao S., Huan Y., Guo F., Guan B., Yang P., Sarao R., Wada T., Leong-Poi H., Crackower M.A., Fukamizu A., Hui C.C., Hein L., Uhlig S., Slutsky A.S., Jiang C., Penninger J.M. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng J.S., Hsu Y.C., Wu H.D., Pan H.Z., Wang H.C., Shun C.T., Yu C.J., Yang P.C. Role of the renin-angiotensin system in ventilator-induced lung injury: an in vivo study in a rat model. Thorax. 2007;62(6):527–535. doi: 10.1136/thx.2006.061945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H.P., Look D.C., Tan P., Shi L., Hickey M., Gakhar L., Chappell M.C., Wohlford-Lenane C., McCray P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297(1):L84–L96. doi: 10.1152/ajplung.00071.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski E.J., Garten R.J., Bridges C.B. Influenza pandemic epidemiologic and virologic diversity: reminding ourselves of the possibilities. Clin. Infect. Dis. 2011;52(Suppl. 1):S44L 49. doi: 10.1093/cid/ciq010. [DOI] [PubMed] [Google Scholar]
- Keidar S., Kaplan M., Gamliel-Lazarovich A. ACE2 of the heart: from angiotensin I to angiotensin (1–7) Cardiovasc. Res. 2007;73(3):463–469. doi: 10.1016/j.cardiores.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Kuba K., Imai Y., Penninger J.M. Angiotensin-converting enzyme 2 in lung diseases. Curr. Opin. Pharmacol. 2006;6(3):271–276. doi: 10.1016/j.coph.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert D.W., Yarski M., Warner F.J., Thornhill P., Parkin E.T., Smith A.I., Hooper N.M., Turner A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J. Biol. Chem. 2005;280(34):30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.H., Goldberg A.L. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- Li W., Greenough T.C., Moore M.J., Vasilieva N., Somasundaran M., Sullivan J.L., Farzan M., Choe H. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin-converting enzyme 2. J. Virol. 2004;78(20):11429–11433. doi: 10.1128/JVI.78.20.11429-11433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N., Murakami A., He Y., Marasco W.A., Guan Y., Choe H., Farzan M. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Molina-Molina M., Abdul-Hafez A., Uhal V., Xaubet A., Uhal B. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;295(1):L178–L185. doi: 10.1152/ajplung.00009.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Molina-Molina M., Abdul-Hafez A., Uhal V., Xaubet A., Uhal B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;295(1):L178–L185. doi: 10.1152/ajplung.00009.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall R.P., Gohlke P., Chambers R.C., Howell D.C., Bottoms S.E., Unger T., McAnulty R.J., Laurent G.J. Angiotensin II and the fibroproliferative response to acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2004;286(1):L156–L164. doi: 10.1152/ajplung.00313.2002. [DOI] [PubMed] [Google Scholar]
- Mitnaul L.J., Castrucci M.R., Murti K.G., Kawaoka Y. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J. Virol. 1996;70(2):873–879. doi: 10.1128/jvi.70.2.873-879.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L., Muench H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938;27(3):493–497. [Google Scholar]
- Suttajit M., Winzler R.J. Effect of modification of N-acetylneuraminic acid on the binding of glycoproteins to influenza virus and on susceptibility to cleavage by neuraminidase. J. Biol. Chem. 1971;246(10):3398–3404. [PubMed] [Google Scholar]
- Tobita K., Sugiura A., Enomote C., Furuyama M. Plaque assay and primary isolation of influenza A viruses in an established line of canine kidney cells (MDCK) in the presence of trypsin. Med. Microbiol. Immunol. 1975;162(1):9–14. doi: 10.1007/BF02123572. [DOI] [PubMed] [Google Scholar]
- Yang N., Hong X., Yang P., Ju X., Wang Y., Tang J., Li C., Fan Q., Zhang F., Chen Z., Xing L., Zhao Z., Gao X., Liao G., Li Q., Wang X., Li D., Jiang C. The 2009 pandemic A/Wenshan/01/2009 H1N1 induces apoptotic cell death in human airway epithelial cells. J. Mol. Cell Biol. 2011;3(4):221–229. doi: 10.1093/jmcb/mjr017. [DOI] [PubMed] [Google Scholar]