











































vasp_tutorial.pptx
- 1. INL/CON-20-59245 Introduction to the Vienna Ab Initio Simulation Package (VASP) Idaho National Laboratory
- 2. INL/CON-20-59245 Online Resources for VASP • John Kitchin’s book: “Modeling materials using density functional theory” (2012) – http://kitchingroup.cheme.cmu.edu/dft-book/dft.html • VASP Workshop Lectures: – https://www.vasp.at/documentation/ • VASP Wiki: – https://www.vasp.at/wiki/index.php/The_VASP_Manual • NERSC VASP Tutorial – https://www.nersc.gov/assets/Uploads/VASP-lecture-Basics.pdf • NERSC Examples for VASP: – https://www.nersc.gov/assets/Uploads/VASP-tutorial- AtomsMoleculesBulk.pdf • Atomic Simulation Environment – https://wiki.fysik.dtu.dk/ase/tutorials/tutorials.html
- 3. INL/CON-20-59245 Density Functional Theory -- Resources DFT from a chemistry perspective: A chemical and theoretical way to look at bonding on surfaces By Ronald Hoffmann https://journals.aps.org/rmp/pdf/10.1103/RevModPhys.60.601 DFT Review Article: Density Functional Theory as a Major Tool in Computational Materials Science By Arthur Freeman https://www.annualreviews.org/doi/10.1146/annurev.ms.25.080195.000255 DFT Implementation details for VASP: Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set By Kresse and Furthmüller https://doi.org/10.1016/0927-0256(96)00008-0
- 4. INL/CON-20-59245 Density Functional Theory -- Background 1926: For a time-independent n-body system: where is the wave function for an n-body system For systems with small number of electrons ( ~O(10) ), solving this is doable. But for larger systems, this becomes too complicated to solve.
- 5. INL/CON-20-59245 Density Functional Theory -- Background Solution: The Hohenberg-Kohn-Sham theory: a systematic way to map the many-body problem onto a single-body problem (Courtesy NERSC) Why is solving the Schrödinger equation for the many-body problem too difficult for anything but a relatively small number of electrons? Storage for example…
- 6. INL/CON-20-59245 Density Functional Theory -- Background What is a functional? Suppose f(x) = x2 ; we call f(x) a function since when we give it a number, it returns a number. A functional returns a number when we give it a function: e.g. g(f(x) is a functional; if I give it f(x) = sin(x), g(f(x)) returns 2.
- 7. INL/CON-20-59245 Density Functional Theory -- Background Walter Kohn (Nobel Prize in Chemistry 1998) The key idea: Don’t rely on the wave function; express material properties (ground state) as a functional of the charge density Why is this important? The charge density is only 3 dimension regardless of the number of electrons! The Hohenberg – Kohn theorems (1964) ensure that the functional exists but doesn’t say how to find it! So the ground state energy of system can be written as: where is some functional of Nobel Lecture: Electronic structure of matter—wave functions and density functionals by W. Kohn https://journals.aps.org/rmp/abstract/10.1103/RevModPhys.71.1253
- 8. INL/CON-20-59245 Density Functional Theory -- Background What is an exchange correlation functional? Generally written in this form: where is the exchange correlation energy density (it represents the energy per electron ) and is the charge density (or number of electrons per unit volume)
- 9. INL/CON-20-59245 Density Functional Theory -- Background There are several types of exchange-correlation functionals: Local Density Approximation (LDA) -- Assumes that the exchange-correlation energy density at every position in space is the same as a uniform electron gas
- 10. INL/CON-20-59245 Density Functional Theory -- Background There are several types of exchange-correlation functionals: Generalized Gradient Approximation (GGA) -- Incorporates the gradients of the charge density at a point as well as the value, i.e. GGA functionals are generally constructed as a correction term, e.g.
- 11. INL/CON-20-59245 Density Functional Theory -- Background There are several types of exchange-correlation functionals: Meta-Generalized Gradient Approximation (GGA) -- Incorporates the Laplacian of the density Hybrid functionals (or Adiabatic Connection Method functionals) -- Include fractions of exact Hartree-Fock exchange energy
- 12. INL/CON-20-59245 Density Functional Theory -- Background Specific exchange-correlation functionals can be selected by setting the GGA flag in the INCAR file of VASP: GGA default is the type of exchange-correlation specified in the POTCAR file Many functionals contains empirical parameters with values fitted to reproduce experiments. Others have no empirically determined parameters (e.g. PW91).
- 13. INL/CON-20-59245 Density Functional Theory -- Background Example: Geometry optimization of water using different exchange correlation functionals From C.K. Skylaris
- 14. INL/CON-20-59245 Density Functional Theory -- Background In VASP the time independent Schrödinger equation becomes where is the eigenvector dependent exchange correlation functional replacing the Hamiltonian operator are the eigenvalues as a function of the eigenvectors or energy bands and the wavefunction is replaced by a radial density expansion that is a superposition of plane waves: where are constants.
- 15. INL/CON-20-59245 Density Functional Theory -- Background Why plane waves? 1. Good choice for systems with repeating structure because it enables a periodic density expansion 2. We can use FFT’s to evaluate the action of the Hamiltonian on the orbitals. FFT’s are O(N log N) and so this approach is computationally compelling. Is VASP the only DFT software that uses plane waves? No. CASTEP also does, among others.
- 16. INL/CON-20-59245 The VASP ENCUT parameter can be used to control this Density Functional Theory -- Background Do we need an infinite number of plane waves? The coefficients of the planewaves ( ) go to zero for high energy planewaves. So we can truncate above a cutoff energy. But the cutoff energy is element dependent! Cutoff energies (eV) for two different elements and two different precision levels. (from John Kitchin)
- 17. INL/CON-20-59245 History of VASP • Based on code written by Mike Payne (then @ MIT) – VASP is similar to CASTEP code because of this (CASTEP = Cambridge Serial Total Energy Package) • 1989 Jürgen Hafner brought the code to U. Vienna • 1993 VASP written by J. Furthmüller and G. Kresse • Development continues under G. Kresse • Stable Releases over the past 10 years: – V6.1.0 28 Jan 2020 – V5.4.4 18 April 2017 – V5.3.3 18 Dec 2012 – V5.2.11 Dec 2010
- 18. INL/CON-20-59245 Pseudopotentials • The core electrons of an atom are computationally expensive with planewave basis sets – Large number of planewaves are required to expand their wavefunctions – Contributions of core electrons versus valence electrons to bonding is usually negligible • To address this, we replace the atomic potential due the to core electrons with a pseudopotential which has the same effect on valence electrons: (see also “Efficient pseudopotentials for plane-wave calculations by Troullier and Martins: – https://journals.aps.org/prb/abstract/10.1103/PhysRevB.43.1993) • VASP provides a database of pseudopotentials based on the Project Augmented-Wave (PAW) method. These are called PAW potentials. – https://journals.aps.org/prb/abstract/10.1103/PhysRevB.50.17953 – https://journals.aps.org/prb/abstract/10.1103/PhysRevB.59.1758
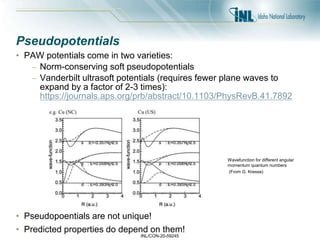
- 19. INL/CON-20-59245 Pseudopotentials • PAW potentials come in two varieties: – Norm-conserving soft pseudopotentials – Vanderbilt ultrasoft potentials (requires fewer plane waves to expand by a factor of 2-3 times): https://journals.aps.org/prb/abstract/10.1103/PhysRevB.41.7892 • Pseudopoentials are not unique! • Predicted properties do depend on them! (From G. Kresse) Wavefunction for different angular momentum quantum numbers
- 20. INL/CON-20-59245 CAR Files • Input and output data in VASP are read from or written to “CAR” files ( a reference to “CARDS” when I/O on computers was done by punchcards • VASP requires at least four CAR files to run: INCAR, POSCAR, POTCAR, and KPOINTS • VASP may also read in: CHGCAR, WAVECAR • VASP will detect the presence of a STOPCAR file • Some VASP output files include: CHG, CHGCAR, CONTCAR, DOSCAR, EIGENVAL, ELFCAR, IBZKPT, LOCPOT, OSZICAR, OUTCAR, PARCHG, PCDAT, PROCAR, WAVECAR,XDATCAR INPUT OUTPUT
- 21. INL/CON-20-59245 INCAR • This is the central input file of VASP. Tip: tabs in this file cause problems with VASP – avoid tabs! Simple Example: IBRION = 2 ISIF = 3 NSW = 100 POTIM = 0.5 ENCUT = 400 PREC = Accurate NBANDS = 20
- 22. INL/CON-20-59245 POSCAR • The “position” card. Contains the lattice geometry and the ion positions. • The first line is a comment line (put your system name here!) • The second line is a scaling factor. • The next three lines are vectors describing the unit cell of the system • Line 6 tells the elements in the system (in order of appearance in the POTCAR file) • Line 7 tells the number of atoms for each atom type • Line 8: May either say “Selective Dynamics” or “Cartesian” or “Direct” coordinates • Next line: positions of the atoms Simple Example:
- 23. INL/CON-20-59245 KPOINTS • The “K” comes from reciprocal space (or k-space). This file specifies the reciprocal lattice for integration in k-space over the first Brillouin zone. Example: the first Brillouin zone of a square lattice
- 24. INL/CON-20-59245 POTCAR • The “Potential” CAR holding the pseudopotentials. Each element has one or more PAW dataset; they can be concatenated together. • VASP comes with a library of PAW datasets. • When you have more than one type of atom, you have to concatenate the PAW datasets IN THE SAME ORDER as atomic species is specified in the POSCAR file. • Each individual PAW data set starts with a descriptive section, specifying amongst other things: – Parameters that were required to generate the dataset: – Number of valence electrons – Atomic mass – Default energy cutoffs
- 26. INL/CON-20-59245 Compiling VASP • Most of the time VASP will already be compiled for you, but it is distributed in source code form so it is likely at some point you will need to compile it. • Compiling VASP requires manual modification of the makefile.include file. • The core computations of VASP are FFT’s and dense linear algebra including eigenvector and eigenvalue computations; hence, the better the BLAS and FFT support you have, the better the performance. • Compiling VASP against OpenBLAS, LAPACK, ScaLAPACK, and FFTW (all open source libraries) is possible and easily done; however, performance is about 3X worse compared with linking against Intel’s MKL libraries (closed, proprietary). • VASP will also compile on ARM architectures; benchmarks show it about 20% slower than Broadwell 22c
- 27. INL/CON-20-59245 VASP on ARM • Cray User 2018 Study on VASP on ARM by Simon McIntosh-Smith: https://uob-hpc.github.io/assets/cug-2018.pdf Calculations by VASP are dominated by floating-point intensive routines which favors x86 processors. ARM gives higher core counts to compensate but benchmarks show VASP about 20% slower than Broadwell and half the speed of Skylake
- 28. INL/CON-20-59245 Profiling VASP • VASP is already fully instrumented for profiling • Add the –DPROFILING flag to the CPP_OPTIONS in makefile.include • Profiling output will be written to the OUTCAR • Profile includes both a flat profile and Cumulative profile
- 29. INL/CON-20-59245 Profiling VASP • You can add custom profiling by encapsulating routines of interest with the API call: PROFILING_START and PROFILING_STOP, e.g. PROFILING_START(‘my_profile_check’) … PROFILING_STOP(‘my_profile_check’)
- 30. INL/CON-20-59245 VASP Executables • There are three executables associated with VASP: – VASP_STD • This is the standard version – for general k-point meshes with collinear spins – VASP_GAM • The “Gamma Point only” version – typical for large unit cells – VASP_NCL • The “non-collinear” version • For general k-point meshes • Used for magnetic structure calculations OR to include spin- orbit interactions in a calculation • Runs 3-D FFT’s on mesh twice the size of VASP_STD
- 31. INL/CON-20-59245 VASP GPU Not all INCAR options are supported in the GPU version (e.g. LREAL = .FALSE. is not supported) • Benchmark comparison (siHugeShort) on Sawtooth: – vasp_gpu on 4 GPUs (one node): 3 min 0 secs – vasp_std on 48 cores (one node): 4 min 32 secs
- 32. INL/CON-20-59245 VASP on Sawtooth @ INL • Sawtooth has a CPU version and a GPU version of VASP • CPU Executable names are: vasp_std, vasp_gam, and vasp_ncl • GPU Executable names: vasp_gpu Note: Not all INCAR options are supported in the GPU version (e.g. LREAL = .FALSE. is not supported) • Benchmark comparison (siHugeShort): – vasp_gpu on 4 GPUs (one node): 3 min 0 secs – vasp_std on 48 cores (one node): 4 min 32 secs
- 33. INL/CON-20-59245 Running VASP in parallel: ways to extract concurrency • VASP has 3 levels of parallelism – Over k-points – Over bands – Over plane-wave coefficients • Related fundamentals: – The number of k-points – The number of bands (determined indirectly by the number of atoms and electrons) – The number of plane waves (which gives the number of grid points in the FFTs)
- 34. INL/CON-20-59245 The Number of Bands • Parallelization over bands is controlled by the NPAR parameter • NPAR determines the number of bands that are treated in parallel • Extreme limit is 1 band per core; this is usually too little work to get good efficiency. • Good rule of thumb: number of bands per core equal to the average number of valence electrons (4-8). • Make sure NPAR evenly divides the number of cores available on a node, e.g.: – On Sawtooth (48 cores/node), best efficiency for NPAR is 6 – On Lemhi (40 cores/node), best efficiency for NPAR is 8 – On Falcon (36 cores/node), best efficiency for NPAR is 6
- 35. INL/CON-20-59245 The Number of K-points • VASP can treat each k-point independently • The number of k-point groups that run concurrently is set by the KPAR parameter • The upper limit for KPAR is the total number of k-points • Larger KPAR values increase the memory overhead • It rarely makes sense to have KPAR larger than the number of compute nodes requested
- 36. INL/CON-20-59245 Basis Set Size • This determines the grid size of the FFTs • 3-D FFT’s in VASP are computed in a plane-wise manner by default (to reduce communication between ranks) • Each of the NPAR groups works on a 2D plane • NGZ is the number of grid points in the Z direction for the FFT • Generally you want NGZ to be evenly divisible by NPAR for best load balancing • You can tune NGZ by adjusting ENCUT or explicitly setting NGZ
- 37. INL/CON-20-59245 Simple Example: Molecular Dynamics run on 32-atom cell • INCAR: # electronic degrees ENCUT = 300 LREAL = A # real space projection PREC = Normal # chose Low only after tests EDIFF = 1E-5 # do not use default (too large drift) ISMEAR = 1 ; SIGMA = 0.1 # Fermi smearing: 2000 K 0.086 10-3 ALGO = Very Fast # recommended for MD (fall back ALGO = Fast) MAXMIX = 40 # reuse mixer from one MD step to next NCORE= 4 # one orbital on 4 cores ISYM = 0 # no symmetry NELMIN = 4 # minimum 4 steps per time step, avoid breaking after 2 steps # MD (do little writing to save disc space) IBRION = 0 ; NSW = 100000 ; NWRITE = 0 ; LCHARG = .FALSE. ; LWAVE = .FALSE. TEBEG = 3063 ; TEEND = 3063 # canonic (Nose) MD with XDATCAR updated every 50 steps
- 38. INL/CON-20-59245 Simple Example: Molecular Dynamics run on 32-atom cell • POSCAR: AgPd (50% Pd) -3% Volume 4.11171 0.00000000 2.00000000 0.00000000 0.00000000 0.00000000 2.00000000 2.00000000 0.00000000 0.00000000 16 16 D 0.00000000 0.00000000 0.00000000 0.25000000 0.00000000 0.25000000 0.25000000 0.25000000 0.00000000 0.00000000 0.50000000 0.00000000 0.25000000 0.50000000 0.25000000 0.75000000 0.25000000 0.00000000 0.00000000 0.00000000 0.50000000 0.25000000 0.25000000 0.50000000 0.00000000 0.75000000 0.75000000 0.25000000 0.75000000 0.50000000
- 39. INL/CON-20-59245 Simple Example: Molecular Dynamics run on 32-atom cell • KPOINTS: Gamma-point only 1 ! one k-point rec ! in units of the reciprocal lattice vector 0 0 0 1 ! 3 coordinates and weight
- 40. INL/CON-20-59245 Some Use Cases for VASP • Calculating surface energies • Calculating formation energies • Calculating reaction energies • Calculating energy barriers – Generally done using the Nudged Elastic Band (NEB) method – VASP Transition State Tools (VTST) assists this • Vibrational Frequency Calculations • Calculating Density of States (DOS) • Calculating band structures -5 -4 -3 -2 -1 0 total s p t2g eg DOS (Courtesy Zongtang Fang)
- 41. INL/CON-20-59245 Tools for VASP VASP Transition State Tools (VTST): http://theory.cm.utexas.edu/vtsttools/ • Provides nudged elastic band (NEB) method implementation for finding saddle points and minimum energy paths • Comes as source code that is added to VASP and then VASP is recompiled
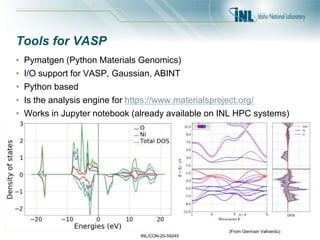
- 42. INL/CON-20-59245 Tools for VASP • Pymatgen (Python Materials Genomics) • I/O support for VASP, Gaussian, ABINT • Python based • Is the analysis engine for https://www.materialsproject.org/ • Works in Jupyter notebook (already available on INL HPC systems) (From Germain Vallverdu)
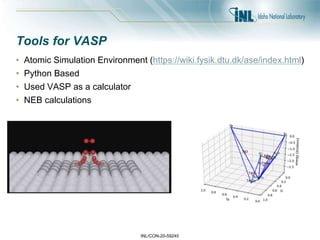
- 43. INL/CON-20-59245 Tools for VASP • Atomic Simulation Environment (https://wiki.fysik.dtu.dk/ase/index.html) • Python Based • Used VASP as a calculator • NEB calculations
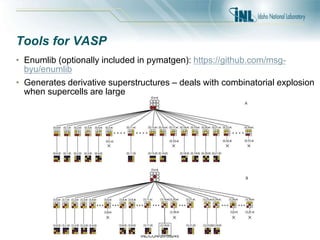
- 44. INL/CON-20-59245 Tools for VASP • Enumlib (optionally included in pymatgen): https://github.com/msg- byu/enumlib • Generates derivative superstructures – deals with combinatorial explosion when supercells are large
- 45. INL/CON-20-59245 VASP6: What’s new? • On-the-fly machine learning for force fields via Bayesian inference • Demonstrated for predicting melting points: – https://journals.aps.org/prb/abstract/10.1103/PhysRevB.100.014105 Large number of machine learning force field (ML FF) parameters added for INCAR, i.e.
- 46. INL/CON-20-59245 VASP6: What’s New? • Faster performance for hybrid DFT calculation • Hybrid OpenMP and MPI parallelization • OpenACC support for GPUs • HDF5 support • Libxc support for gradient correct functionals • Time evolution support for frequency dependent response functions
- 47. INL/CON-20-59245 Machine Learning Opportunities for VASP • Neural network architectures for modeling atomic systems – Quantum-chemical insights from deep tensor neural networks https://www.nature.com/articles/ncomms13890 – A deep learning Toolbox for atomistic systems https://pubs.acs.org/doi/10.1021/acs.jctc.8b00908 – Transfer Learning for material properties https://arxiv.org/pdf/2006.16420.pdf • Databases from high-throughput ab initio calculations for training like AFLOWLIB: https://doi.org/10.1016/j.commatsci.2012.02.002 • On-the-fly Learning: On-the-fly machine learning force field generation: Application to melting points – https://journals.aps.org/prb/abstract/10.1103/PhysRevB.100.014105
- 48. INL/CON-20-59245 Some Resources for VASP • VEST: http://jp-minerals.org/vesta/en/ – Free crystal structure viewer and building. Can read and write POSCAR and CONTCAR files; can read CHGCAR, PARCHG, LOCPOT, and ELFCAR. • RINGS: http://rings-code.sourceforge.net/ – Extracts pair distribution functions • VTST Tools: http://theory.cm.utexas.edu/vtsttools/ – Transition state tools for finding saddle points. • VMD: http://www.uni-due.de/~hp0058/?file=vmdplugins.html&lang=en. – Used to visualize structures and trajectories • CIF2Cell: http://sourceforge.net/projects/cif2cell – Tool to generate a POSCAR from a Crystallographic Information Framework (CIF) file. • Atomic Simulation Environment: https://wiki.fysik.dtu.dk/ase/ • Pymatgen: https://pymatgen.org/index.html • Others: https://www.vasp.at/index.php/resources