Use of 3D QSAR Models For Database Screening: A Feasibility Study
Use of 3D QSAR Models For Database Screening: A Feasibility Study
Download as pdf or txt
You might also like
- 85.3 % Natural Origin : Moisturizing Lip InkDocument2 pages85.3 % Natural Origin : Moisturizing Lip InkDanyal2222100% (1)
- Household Toilet Bowl Cleaner Formulations: E - TextbooksDocument14 pagesHousehold Toilet Bowl Cleaner Formulations: E - TextbooksDanyal2222100% (3)
- US2565067Document3 pagesUS2565067Danyal2222No ratings yet
- Tobioij 7 63Document5 pagesTobioij 7 63Heneampong IsaacNo ratings yet
- Three-Dimensional Classification Structure-Activity Relationship Analysis Using Convolutional Neural NetworkDocument8 pagesThree-Dimensional Classification Structure-Activity Relationship Analysis Using Convolutional Neural NetworkWinwin Karunia DiningsihNo ratings yet
- Medicinal Chemistry and The Molecular Operating Environment (MOE) : Application of QSAR and Molecular Docking To Drug DiscoveryDocument18 pagesMedicinal Chemistry and The Molecular Operating Environment (MOE) : Application of QSAR and Molecular Docking To Drug Discoverydwi koko pratokoNo ratings yet
- Quantitative Structure-Activity RelationshipDocument11 pagesQuantitative Structure-Activity RelationshipfaridsidikNo ratings yet
- 2d and 3d QsarDocument15 pages2d and 3d QsarBinh CaoNo ratings yet
- Application of GA-MLR Method in QSPR Modeling of S PDFDocument11 pagesApplication of GA-MLR Method in QSPR Modeling of S PDFTientcheu FrankNo ratings yet
- Qsar New 1Document21 pagesQsar New 1Heneampong IsaacNo ratings yet
- Cross Validation in Stochastic Analytic ContinuationDocument18 pagesCross Validation in Stochastic Analytic ContinuationxoqwfifhuzkxlztmxeNo ratings yet
- Descritores e Seus Métodos de Seleção Na Análise QSAR: Paradigma para o Design de MedicamentosDocument12 pagesDescritores e Seus Métodos de Seleção Na Análise QSAR: Paradigma para o Design de MedicamentosLucas PalmeiraNo ratings yet
- Boinformatic SmellDocument5 pagesBoinformatic SmellJessica SofiattiNo ratings yet
- Comfa and Related ApproachesDocument23 pagesComfa and Related ApproachesLina Rodriguez AponteNo ratings yet
- Interpretation of Quantitative Structure-Property and - Activity RelationshipsDocument7 pagesInterpretation of Quantitative Structure-Property and - Activity RelationshipsreilyshawnNo ratings yet
- Bacilieri&Moro, 2006 - Ligand-Based Drug Design Methodologies in Drug Discovery Process An OverviewDocument12 pagesBacilieri&Moro, 2006 - Ligand-Based Drug Design Methodologies in Drug Discovery Process An OverviewDiana López LópezNo ratings yet
- 11 - Chapter 5 PDFDocument59 pages11 - Chapter 5 PDFTukai KulkarniNo ratings yet
- International Journal of Research and Development in Pharmacy and Life SciencesDocument9 pagesInternational Journal of Research and Development in Pharmacy and Life SciencesAkash NarayananNo ratings yet
- A Novel Remaining Useful Life Prediction Method FoDocument14 pagesA Novel Remaining Useful Life Prediction Method FoKeren Persis PNo ratings yet
- Text Kuliah Metoda Statistika QSAR KIMED 2023 PDFDocument22 pagesText Kuliah Metoda Statistika QSAR KIMED 2023 PDFameljpNo ratings yet
- Prediction Reliability of QSAR Models AnDocument17 pagesPrediction Reliability of QSAR Models AndrfperezNo ratings yet
- 15 Chapter 6Document26 pages15 Chapter 6NeelutpalGogoiNo ratings yet
- Processes-11-03325 Machine LearningDocument40 pagesProcesses-11-03325 Machine LearningFrank MtetwaNo ratings yet
- An Atomistic Machine Learning Package For Surface ScienceDocument42 pagesAn Atomistic Machine Learning Package For Surface Sciencebmalki68No ratings yet
- 11 Vol 14 Issue 3 March 2023 IJPSR RE 5435Document18 pages11 Vol 14 Issue 3 March 2023 IJPSR RE 5435Yem ConcepcionNo ratings yet
- Estimation of Properties of Homologous Series With Targeted Quantitative Structure-Property RelationshipsDocument11 pagesEstimation of Properties of Homologous Series With Targeted Quantitative Structure-Property Relationshipsvu anh ducNo ratings yet
- .Document10 pages.Trần Duy TânNo ratings yet
- Clustering Molecular Dynamics Trajectories - I. Characterizing The Performance of Different Clustering AlgorithmsDocument44 pagesClustering Molecular Dynamics Trajectories - I. Characterizing The Performance of Different Clustering AlgorithmsyunniemNo ratings yet
- A Three-Stage Automated Modal Identification Framework For BridgeDocument14 pagesA Three-Stage Automated Modal Identification Framework For Bridgezhengyifu138No ratings yet
- 2013 On Exploring Structure Activity RelationshipsDocument14 pages2013 On Exploring Structure Activity RelationshipsAura RusuNo ratings yet
- Representing Local Atomic Environment Using Descriptors Based On Local CorrelationsDocument17 pagesRepresenting Local Atomic Environment Using Descriptors Based On Local CorrelationsSuryaKaushik . Tutorials and projectsNo ratings yet
- In SilicoDocument14 pagesIn Silicoibro4khadNo ratings yet
- 10 21105 Joss 05057Document5 pages10 21105 Joss 05057Arpanet ArpanetNo ratings yet
- Two Personal Perspectives On A Key Issue Comtemporary 3D QSAR, Clark and Norinder, 20may12Document6 pagesTwo Personal Perspectives On A Key Issue Comtemporary 3D QSAR, Clark and Norinder, 20may12Vida Faith GalvezNo ratings yet
- Dock Around The Clock - Current Status of Small MoleculeDocument11 pagesDock Around The Clock - Current Status of Small Moleculetaoufik akabliNo ratings yet
- DatabasesDocument4 pagesDatabasesmeenujmsNo ratings yet
- Automated Docking Using A LGADocument24 pagesAutomated Docking Using A LGAtaoufik akabliNo ratings yet
- Research Papers On Qsar StudiesDocument7 pagesResearch Papers On Qsar Studiesgz8reqdc100% (1)
- Video 19Document31 pagesVideo 19kamutmazNo ratings yet
- A Concise Review On Recent Developments of Machine Learning For The Prediction of Vibrational Spectra-NADocument12 pagesA Concise Review On Recent Developments of Machine Learning For The Prediction of Vibrational Spectra-NAalizahid000No ratings yet
- Improved Prediction of Protein Side-Chain Conformations With SCWRL4Document18 pagesImproved Prediction of Protein Side-Chain Conformations With SCWRL4dunbrackNo ratings yet
- Ann 3Document8 pagesAnn 3Saul Antonio Montoya SerranoNo ratings yet
- Predictive Model For The Design of Zwitterionic Polymer Brushes: A Statistical Design of Experiments ApproachDocument9 pagesPredictive Model For The Design of Zwitterionic Polymer Brushes: A Statistical Design of Experiments ApproachrmykmrlvNo ratings yet
- Renewable and Sustainable Energy Reviews: Hegazy Rezk, Ahmed Fathy, Almoataz Y. AbdelazizDocument10 pagesRenewable and Sustainable Energy Reviews: Hegazy Rezk, Ahmed Fathy, Almoataz Y. AbdelazizDany ApablazaNo ratings yet
- Acssynbio 9b00523Document14 pagesAcssynbio 9b00523PrinsNo ratings yet
- ACCESS3277543Document18 pagesACCESS3277543Doomed DarwinNo ratings yet
- Parameter Estimation With Term-Wise Decomposition in Biochemical Network GMA Models by Hybrid Regularized Least Squares-Particle Swarm OptimizationDocument8 pagesParameter Estimation With Term-Wise Decomposition in Biochemical Network GMA Models by Hybrid Regularized Least Squares-Particle Swarm OptimizationGek CagatanNo ratings yet
- Estimation of Cavitation Velocity Fields Based On Limited Pressure Data Through Improved U-Shaped Neural NetworkDocument22 pagesEstimation of Cavitation Velocity Fields Based On Limited Pressure Data Through Improved U-Shaped Neural Networkzhangzhigu9No ratings yet
- Active Site Representation in First-Principles Microkinetic ModelsDocument8 pagesActive Site Representation in First-Principles Microkinetic Modelsrevolvevijaya123No ratings yet
- Robust Principal Component Analysis For Modal Decomposition of Corrupt Fluid FlowsDocument24 pagesRobust Principal Component Analysis For Modal Decomposition of Corrupt Fluid Flows吴吉人No ratings yet
- Machine Learning in RNA Structure Prediction - Advances and ChallengesDocument11 pagesMachine Learning in RNA Structure Prediction - Advances and Challengesyahel.godinezNo ratings yet
- Autocorrelation Descriptor Improvements For QSAR - 2DA - Sign and 3DA - SignDocument9 pagesAutocorrelation Descriptor Improvements For QSAR - 2DA - Sign and 3DA - SignRadite YogaswaraNo ratings yet
- Machine Learning Based Power Estimation For CMOS VDocument12 pagesMachine Learning Based Power Estimation For CMOS Vsohan kambleNo ratings yet
- Self-Organizing Neural Network For Modeling 3D QSAR of ColchicinoidsDocument9 pagesSelf-Organizing Neural Network For Modeling 3D QSAR of ColchicinoidsAnonymous S6SrhtNo ratings yet
- QSARDocument35 pagesQSARSathish Kumar100% (2)
- Balaji and Brake - 2019 - The Surrogate System Hypothesis For Joint MechanicDocument23 pagesBalaji and Brake - 2019 - The Surrogate System Hypothesis For Joint MechanicNidish NarayanaaNo ratings yet
- A Data Driven Probabilistic Framework Towards The in Situ Prognostics of Fatigue Life of Composites Based On Acoustic Emission Data PDFDocument8 pagesA Data Driven Probabilistic Framework Towards The in Situ Prognostics of Fatigue Life of Composites Based On Acoustic Emission Data PDFHanifNo ratings yet
- Multi-Task Neural Networks For QSAR PredictionsDocument21 pagesMulti-Task Neural Networks For QSAR PredictionsKunel KumarNo ratings yet
- IJAIADocument8 pagesIJAIAAdam HansenNo ratings yet
- 2022 Interpretable Machine Learning For KnowledgeDocument10 pages2022 Interpretable Machine Learning For KnowledgekeyachemNo ratings yet
- Data Based ModellingDocument20 pagesData Based ModellingsoumikfarhanNo ratings yet
- Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug DiscoveryDocument16 pagesLarge Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug DiscoverydashanunravanNo ratings yet
- Mechanical Properties of Nanostructured Materials: Quantum Mechanics and Molecular Dynamics InsightsFrom EverandMechanical Properties of Nanostructured Materials: Quantum Mechanics and Molecular Dynamics InsightsNo ratings yet
- Glossy Glide: For Smooth Glide and Instant GlossDocument2 pagesGlossy Glide: For Smooth Glide and Instant GlossDanyal2222No ratings yet
- Book of Proceedings EsdAveiro2021 OnlineDocument302 pagesBook of Proceedings EsdAveiro2021 OnlineDanyal2222No ratings yet
- Tergitol np9 Surfactant TdsDocument2 pagesTergitol np9 Surfactant TdsDanyal2222No ratings yet
- Skincreamhgfactsheet May31 FinalDocument4 pagesSkincreamhgfactsheet May31 FinalDanyal2222No ratings yet
- Isiri-Iso-Iec 17025Document53 pagesIsiri-Iso-Iec 17025Danyal2222No ratings yet
- Theuns 1986Document13 pagesTheuns 1986Danyal2222No ratings yet
- Topical Acne Drug Products For Over The Counter Human Use Revision of Labeling and Classification of Benzoyl Peroxide As Safe and EffectiveDocument11 pagesTopical Acne Drug Products For Over The Counter Human Use Revision of Labeling and Classification of Benzoyl Peroxide As Safe and EffectiveDanyal2222No ratings yet
- Sa enDocument1 pageSa enDanyal2222No ratings yet
- Titration of Anionic Surfactants: DescriptionDocument5 pagesTitration of Anionic Surfactants: DescriptionDanyal2222No ratings yet
- Sikalastomer 710Document1 pageSikalastomer 710Eliza ManNo ratings yet
- Poonam 2019Document25 pagesPoonam 2019RajuNo ratings yet
- ID Kajian Kesuburan Tanah Di Desa Sihiong SDocument10 pagesID Kajian Kesuburan Tanah Di Desa Sihiong SMittaNo ratings yet
- Distillation: Basic ProcedureDocument6 pagesDistillation: Basic Procedurezzrot1No ratings yet
- Exer 10Document7 pagesExer 10Yana CatherineNo ratings yet
- Effect of High Pressure On Fish Meat Quality e A ReviewDocument19 pagesEffect of High Pressure On Fish Meat Quality e A Reviewgs64569No ratings yet
- TC - WIRE Type KDocument1 pageTC - WIRE Type KHou YangfanNo ratings yet
- Painting ProcedureDocument20 pagesPainting ProcedureCRUSTEK ENGINEERSNo ratings yet
- Instruction Manuals - Parr Instrument CompanyDocument7 pagesInstruction Manuals - Parr Instrument CompanyMika SuominenNo ratings yet
- Pointe Chemwell 2910 PDFDocument29 pagesPointe Chemwell 2910 PDFRuben DuranNo ratings yet
- Classification of ElementsDocument14 pagesClassification of Elementskrishna priyaNo ratings yet
- Master'S Degree Programme in Advanced Spectroscopy in ChemistryDocument6 pagesMaster'S Degree Programme in Advanced Spectroscopy in ChemistryHema VarthiniNo ratings yet
- 1 s2.0 S0008622321005674 main 导热吸波BNDocument10 pages1 s2.0 S0008622321005674 main 导热吸波BNVan HenryNo ratings yet
- WaterDocument16 pagesWaterReza Febryantara100% (1)
- DistillationDocument80 pagesDistillationMatewos SadaNo ratings yet
- Module 4: Alkenes: Nomenclature, Preparation and ReactionDocument2 pagesModule 4: Alkenes: Nomenclature, Preparation and ReactionPeña, Rodolfo100% (1)
- Application Li Batteries 5110 Icp-Oes 5994-1937en Us AgilentDocument4 pagesApplication Li Batteries 5110 Icp-Oes 5994-1937en Us AgilentxxxNo ratings yet
- HF-7 Rudder CarrierDocument22 pagesHF-7 Rudder CarrierBorysNo ratings yet
- Preparation of Chalcones: Experiment 4Document4 pagesPreparation of Chalcones: Experiment 4Victor SimonNo ratings yet
- Pyrolysis of Wood-Biomass For Bio-Oil A Critical Review-2Document9 pagesPyrolysis of Wood-Biomass For Bio-Oil A Critical Review-2Some ParawhoreNo ratings yet
- Synthesis and Characterization of Cellulose PhosphateDocument29 pagesSynthesis and Characterization of Cellulose PhosphateEju roslanNo ratings yet
- Flashcards - Topic 5 Electricity and Chemistry - CIE Chemistry IGCSEDocument65 pagesFlashcards - Topic 5 Electricity and Chemistry - CIE Chemistry IGCSEBhawana SinghNo ratings yet
- Chem Ia Exemplar2Document12 pagesChem Ia Exemplar2Archit GargNo ratings yet
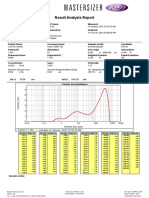
- Result Analysis Report: Um D (0.9) : 141.732 464.814 D (0.1) : Um Um 4.270 D (0.5)Document1 pageResult Analysis Report: Um D (0.9) : 141.732 464.814 D (0.1) : Um Um 4.270 D (0.5)Baher SaidNo ratings yet
- Amat 2010 - Investigation of Spectroscopic Properties of Indigo BlueDocument9 pagesAmat 2010 - Investigation of Spectroscopic Properties of Indigo BlueKaterina MarnellouNo ratings yet
- Part2-Phase Rule-Lecture 6 PDFDocument13 pagesPart2-Phase Rule-Lecture 6 PDFshahad KNo ratings yet
- BSP12.STD.01.01 - Pipe SupportDocument40 pagesBSP12.STD.01.01 - Pipe SupportVINOTH MECHNo ratings yet
- Microleakage PDFDocument16 pagesMicroleakage PDFbyyou0% (1)
- Polymer Clay NanocompositesDocument6 pagesPolymer Clay Nanocompositeshamidrezaee008No ratings yet
- Module-2 Corrosion and Metal Finishing: Sub:Engg ChemistryDocument12 pagesModule-2 Corrosion and Metal Finishing: Sub:Engg Chemistrygani prasadNo ratings yet