

Crystal structure prediction using neural network potential and age-fitness Pareto genetic algorithm


Abstract
While crystal structure prediction (CSP) remains a longstanding challenge, we introduce ParetoCSP, a novel algorithm for CSP, which combines a multi-objective genetic algorithm (GA) with a neural network inter-atomic potential model to find energetically optimal crystal structures given chemical compositions. We enhance the updated multi-objective GA (NSGA-III) by incorporating the genotypic age as an independent optimization criterion and employ the M3GNet universal inter-atomic potential to guide the GA search. Compared to GN-OA, a state-of-the-art neural potential-based CSP algorithm, ParetoCSP demonstrated significantly better predictive capabilities, outperforming by a factor of 2.562 across 55 diverse benchmark structures, as evaluated by seven performance metrics. Trajectory analysis of the traversed structures of all algorithms shows that ParetoCSP generated more valid structures than other algorithms, which helped guide the GA to search more effectively for the optimal structures. Our implementation code is available at https://github.com/sadmanomee/ParetoCSP.
Keywords
INTRODUCTION
Crystal structure prediction (CSP) is the problem of predicting the most energetically stable structure of a crystal given its chemical composition. Knowing the atomic structure is the most crucial aspect of comprehending crystalline materials. With the structural information of the material, advanced quantum-mechanical methods such as Density Functional Theory (DFT) can be utilized to calculate numerous physical characteristics of the crystal[1]. As the physical and chemical characteristics of a crystal are dictated by the arrangement and composition of its atoms, CSP is critical to finding new materials that possess the needed properties such as high thermal conductivity, high compressing strength, high electrical conductivity, or low refractive index. CSP-based computational materials discovery is significant and has the potential to revolutionize a range of industries, such as those involving electric vehicles, Li batteries, building construction, energy storage, and quantum computing hardware [2–6]. For this reason, CSP, along with machine learning (ML)-based inverse design [5,7–10], has emerged as one of the most potential methods for finding novel materials.
Although there have been notable advancements in the field of CSP, the scientific community has yet to solve this fundamental challenge that has persisted for decades. CSP presents a significant challenge due to the requirement to search through an extensive range of potential configurations to identify the most stable arrangement of atoms of a crystal in a high-dimensional space. The complexity of CSP stems from the combinatorial nature of the optimization challenge, where the number of potential configurations grows exponentially with the number of atoms present in the crystal [1]. Additionally, the prediction of the most stable structure relies on several factors, including temperature, pressure, and chemical composition, further increasing the intricacy of the problem. Historically, the main method for determining crystal structures was through experimental X-ray diffraction (XRD) [11], which is time-consuming, expensive, and sometimes impossible, particularly for materials that are difficult to synthesize.
Computational approaches for CSP provide a faster and more affordable alternative than experimental methods. A typical strategy involves searching for the crystal's lowest energy atomic arrangement by optimizing its potential energy surface (PES) using different search algorithms. However, in some cases, simpler metrics such as the cohesive energy or the formation energy of the structures can be used instead [4]. The highly non-convex nature of the PES, which can contain a vast number of local minima, reduces the efficiency of the search algorithms. Moreover, finding the global minimum of a PES is categorized as an NP-hard problem [12]. Most research on the CSP problem concentrates on ab initio techniques, which involve exploring the atomic configuration space to locate the most stable structure based on the first-principles calculations of the free energy of possible structures [13–15]. Although these methods are highly accurate, the scalability and the applicability of these ab initio algorithms for predicting crystal structures remain a challenge. These methods are severely constrained because they rely on expensive first-principles DFT calculations [16,17] to determine the free energy of candidate structures. Furthermore, these methods are only applicable for predicting structures of comparatively small systems (< 10-20 atoms in the unit cell). Although there are inexpensive models available to estimate the free energy, they tend to have a poor correlation with reality, which can result in an inaccurate search [14]. For example, state-of-the-art (SOTA) graph neural networks (GNNs) have demonstrated the capability to accurately predict the formation energy of candidate structures [18–23], their performance on predicting non-stable or meta-stable structures is significantly lower as they are usually trained with stable crystals.
Several search algorithms have been applied to the CSP problem, including random sampling [12], simulated annealing [24–26], meta-dynamics [27,28], basin hopping [29,30], minima hopping [31], genetic algorithm (GA) [14,32–34], particle swarm optimization (PSO) [15], Bayesian optimization (BO) [35,36], and deep learning (DL) [37,38]. Among them, the USPEX (Universal Structure Predictor: Evolutionary Xtallography) algorithm, developed by Glass et al., is a prominent CSP algorithm based on evolutionary principles, using natural selection and reproduction to generate new crystal structures [14]. It incorporates a combination of three operators - heredity, mutation, and permutation to explore the configuration space. To evaluate candidate structures, they use ab initio free energy calculation using tools such as VASP [39] and SIESTA [40] which are highly accurate but extremely time-consuming. Another important CSP algorithm named CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) was devised by Wang et al., which employs a PSO algorithm to explore the energy landscape of crystal structures and identify the lowest energy structures [15]. To accomplish this, they developed a special strategy for removing comparable structures and applied symmetry-breaking restrictions to boost search effectiveness. Both USPEX and CALYPSO methods have been successfully applied to predict the crystal structures of diverse materials, including those under high-pressure conditions, complex oxides, alloys, etc. The random sampling-based CSP algorithms have also demonstrated their effectiveness. For example, AIRSS (Ab Initio Random Structure Searching) presented by Pickard et al. describes a scheme that generates diverse random crystal structures for various types of crystals and conducts DFT calculations on them to determine the most stable one[12]. Another genre of CSP methods are template-based approaches [41–43], which involves finding an existing crystal structure as the template using some heuristic methods, or the ML method, etc., which has a similar chemical formula and then replacing some of its atoms with different elements. However, the accuracy of these models is constrained by the diversity and availability of the templates and the complexity of the target compound. Inspired by the recent success of DL-based methods in protein structure prediction [44–46], a DL-based algorithm, AlphaCrystal [38], has been designed to predict the contact map of a target crystal and then reconstruct its structure via a GA. However, the effectiveness of this model is constrained because its performance relies on the accuracy of the predicted space group, lattice parameters, and distance matrices. Moreover, it ultimately depends on the optimization algorithm for reconstructing the final structure from the contact map as it is unable to provide end-to-end prediction akin to DeepMind's AlphaFold
Compared to previous DFT-based CSP algorithms such as USPEX and CALYPSO, a major progress in CSP is to use ML potential models to replace the costly first-principle energy calculation. Cheng et al. developed a CSP framework named GN-OA, in which a GNN model was first trained to predict the formation energy and then an optimization algorithm was then used to search for the crystal structure with the minimum formation energy, guided by the GNN energy model [36]. They show that the BO search algorithm produces the best results among all optimization algorithms. However, predicting formation energy using GNNs has its drawbacks as its performance largely depends on the dataset it is trained on. A structure search trajectory analysis [47] also showed that current BO and PSO in GN-OA tend to generate too many invalid structures, which deteriorates its performance. While both USPEX and CALYPSO have been combined with ML potentials for CSP before GN-OA, they were only applicable to small crystal systems such as Carbon structures, Sodium under pressure, and Boron clusters [48,49] due to the limitation of their ML potential models. Recently, significant progress has been achieved in ML potentials for crystals [50–54] that can work with multi-element crystals and larger crystals systems. This will bring unprecedented opportunities and promise for modern CSP research and materials discovery. For example, recent advancement in deep neural network-based energy potential [M3GNet IAP (inter-atomic potential)][53] has shown its capability to cover
Our contribution in this paper can be summarized as follows:
● We develop an efficient ParetoCSP for CSP, which combines an updated multi-objective GA (NSGA-III) by the inclusion of the age fitness Pareto optimization criterion and a neural network potential (M3GNet IAP), utilized to correlate crystal structures to their final energy.
● Our systematic evaluations on 55 benchmark crystals show that ParetoCSP outperforms GN-OA by a factor of 2.562 in terms of prediction accuracy.
● We reinforce GN-OA by replacing its formation energy predictor MEGNet (MatErials Graph Network) [19] with the M3GNet IAP final energy model and show that it improves the default GN-OA by a factor of 1.5 in terms of prediction accuracy. We further demonstrated the significant improvement in the search capability of ParetoCSP by showing that ParetoCSP outperforms the updated GN-OA by a factor of 1.71 in terms of prediction accuracy.
● We provide a quantitative analysis of the structures generated by ParetoCSP using seven performance metrics and empirically show that ParetoCSP found better quality structures for the test formulas than those by GN-OA.
● We perform a trajectory analysis of the generated structures by all evaluated CSP algorithms and show that ParetoCSP generates a significantly greater number of valid solutions than the GN-OA algorithm, which may have contributed to ParetoCSP's better performance in predicting the crystal structures.
METHOD
ParetoCSP: algorithm description
The input of our algorithm (ParetoCSP) is the elemental composition of a crystal
Our algorithm is based on the idea of the GN-OA algorithm [36] with two major upgrades, including the multi-objective GA search algorithm and the use of M3GNet potential for energy calculation. GN-OA has been proven from previous research that incorporating symmetry constraint expedites CSP [36,55]. Similar to the GN-OA approach, our method also considers CSP with symmetry constraints. We incorporate two additional structural features, namely crystal symmetry
By selecting different combinations of
First, we generate
Next, we perform the genetic operations - crossover and mutation. After crossover, we update the age of each individual by inheriting the maximum age of corresponding parents. Similarly, after mutation, individual ages are updated by inheriting the age of their respective parents. These operations result in a new population of

Figure 1. The flowchart of ParetoCSP algorithm. It starts by generating
AFPO
One of the key requirements for a GA to achieve a robust global search is to maintain the diversity of the population. Here, we employed the multi-objective GA, AFPO by Schmidt and Lipson [59], to achieve this goal. The AFPO algorithm is inspired by the idea of age-layered population structure (ALPS) [63,64], which divides the evolving population into layers based on how long the genetic material has been present in the population so that competitions happen at different fitness levels, avoiding the occurrence of premature convergence. The age of an individual is defined as how long the oldest part of its genotype has been present in the population [65]. Instead of partitioning the population into layers as done in the Hierarchical Fair Competition (HFC) algorithm [63], AFPO uses age as an explicit optimization criterion (an independent dimension in a multi-objective Pareto front). A solution is considered optimal if it has both higher fitness and lower age compared to others. This enables the algorithm to maintain diversity in the population, avoid premature convergence to local optima, and find better solutions at faster convergence speed [59]. The AFPO algorithm starts by initializing a population of
NSGA-III: multi-objective GA
We use the NSGA-III [57] algorithm to implement the age-fitness-based GA AFPO. NSGA-III is an improved version of the popular multi-objective evolutionary algorithm NSGA-II [66]. Here, we describe the NSGA-III framework as defined in reference [57,58]. The NSGA-III algorithm begins with defining a group of reference points. To create an offspring population
The selection process of NSGA-III is substantially altered from the approach used in NSGA-II. First, the objective values and reference points are normalized. Second, each member in
M3GNet inter-atomic potential
Energy potential is one of the key components of modern CSP algorithms. Here, we use M3GNet [53], which is a GNN-based ML potential model that explicitly incorporates
In the M3GNet model, position-included graphs serve as inputs. Graph features include embedded atomic numbers of elements and pair bond distances. Similar to traditional GNNs, the node and the edge features are updated via the graph convolution operations. Architecturally, there are several differences between M3GNet and MEGNet. For example, M3GNet utilizes angle information between bonds, but MEGNet does not. Unlike MEGNet, M3GNet has the many-body computation module that was proven essential for its excellent performance. Additionally, M3GNet uses gated multi-layer perceptron (MLP) for obtaining atomic energy in contrast to MEGNet. Our M3GNet potential was trained using both stable and unstable structures so that it can well capture the difference between these two. For details about the training, we refer the readers to the M3GNet paper [53]. The dataset used for training the M3GNet model is much larger than that for the MEGNet model, and it contains both stable and unstable structures which makes M3GNet more specialized in predicting energies of the intermediate unstable structures of crystals. The precise and efficient relaxation of diverse crystal structures and the accurate energy prediction achieved by the M3GNet-based relaxation algorithm make it well-suited for large-scale and fast CSP.
Evaluation criteria
Many earlier studies [12,14,15] have depended on manual structural examination and ab initio formation energy comparison to assess the performance of a CSP algorithm. However, these metrics do not address the situation that an algorithm may not find the exact solution for a crystal, and it is not clear how much the generated structure deviates from the ground truth structure. Usually, previous works did not quantitatively report how good or bad a solution is. Also, if two algorithms fail to generate the exact crystal structure, these metrics do not describe which one is closer to finding the optimal solution. Recently, Wei et al. proposed a set of performance metrics to measure CSP performance which alleviated this issue greatly [47]. We used seven performance metrics from that work to measure the performance of our CSP algorithm and the baselines. The required data are the crystallographic information file (CIF) of both the optimized and relaxed final structure generated by the CSP algorithm and its corresponding ground truth stable structure. Details about these performance metrics can be found in ref[47]. They are shortly listed below:
1. Energy distance (ED)
2. Wyckoff position fraction coordinate root mean squared error distance (W
3. Wyckoff position fraction coordinate root mean absolute error (W
4. Sinkhorn distance (SD)
5. Chamfer distance (CD)
6. Hausdorff distance (HD)
7. Crystal fingerprint distance (FP)
RESULTS
Our objective is to demonstrate the effectiveness of ParetoCSP for CSP by showing that the multi-objective AFPO GA enables a much more effective structure search method than the BO and PSO and that M3GNet IAP is a more powerful crystal energy predictor than the previous MEGNet model.
Benchmark set description
We selected a diverse set of
Details of the
| Composition | No. ofatoms | Space group | Formation energy(eV/atom) | Final energy(eV/atom) | M3GNet final energy(eV/atom) |
| The first | |||||
| TiCo | |||||
| CrPd | |||||
| GaNi | |||||
| ZrSe | |||||
| MnAl | |||||
| NiS | |||||
| TiO | |||||
| NiCl | |||||
| AlNi | |||||
| CuBr | |||||
| VPt | |||||
| MnCo | |||||
| BN | |||||
| GeMo | |||||
| Ca | |||||
| Ga | |||||
| CoAs | |||||
| Li | |||||
| VS | |||||
| Ba | |||||
| SrTiO | |||||
| Al | |||||
| GaBN | |||||
| AcMnO | |||||
| PaTlO | |||||
| CdCuN | |||||
| HoHSe | |||||
| Li | |||||
| Cd | |||||
| AlCrFe | |||||
| ZnCdPt | |||||
| EuAlSi | |||||
| Sc | |||||
| GaSeCl | |||||
| CaAgN | |||||
| BaAlGe | |||||
| K | |||||
| KCrO | |||||
| TiZnCu | |||||
| Ta | |||||
| AgBiSeS | |||||
| ZrTaNO | |||||
| MnAlCuPd | |||||
| CsNaICl | |||||
| DyThCN | |||||
| Li | |||||
| SrWNO | |||||
| Sr | |||||
| ZrCuSiAs | |||||
| NdNiSnH | |||||
| MnCoSnRh | |||||
| Mg | |||||
| AlCr | |||||
| Y | |||||
| Ba | |||||
Performance analysis of ParetoCSP
The default version of ParetoCSP uses M3GNet universal IAP as the final energy evaluator for the candidate structures to guide the AFPO-based GA in identifying the most stable structure with the minimum energy. Our algorithm ParetoCSP predicted the exact structures for
Performance comparison of ParetoCSP with baseline algorithms
| Composition | ParetoCSPwith M3GNet (Default) | ParetoCSPwith MEGNet | GN-OAwith M3GNet | GN-OAwith MEGNet (Default) |
| Successful and failed predictions via manual inspection are denoted by a ✔ and ✘, respectively. ParetoCSP with M3GNet achieved the highest success rate in finding the exact structures of these crystals; GN-OA with M3GNet achieved the second best success rate. ParetoCSP with MEGNet performed as the third-best, while GN-OA with MEGNet performed the poorest. These results highlight the significant impact of using M3GNet IAP as a crystal final energy predictor and structure relaxer and the effectiveness of the AFPO-based GA as a structure search function.AFPO: age-fitness Pareto optimization. | ||||
| TiCo | ✔ | ✔ | ✔ | ✘ |
| CrPd | ✔ | ✔ | ✘ | ✘ |
| GaNi | ✔ | ✔ | ✔ | ✔ |
| ZrSe | ✔ | ✔ | ✔ | ✔ |
| MnAl | ✔ | ✔ | ✔ | ✔ |
| NiS | ✔ | ✘ | ✔ | ✔ |
| TiO | ✔ | ✔ | ✔ | ✔ |
| NiCl | ✔ | ✘ | ✘ | ✘ |
| AlNi | ✔ | ✔ | ✔ | ✔ |
| CuBr | ✔ | ✘ | ✘ | ✘ |
| VPt | ✔ | ✔ | ✔ | ✔ |
| MnCo | ✔ | ✔ | ✔ | ✔ |
| BN | ✔ | ✔ | ✔ | ✔ |
| GeMo | ✔ | ✔ | ✔ | ✔ |
| Ca | ✔ | ✔ | ✘ | ✘ |
| Ga | ✘ | ✘ | ✘ | ✘ |
| CoAs | ✔ | ✘ | ✘ | ✘ |
| Li | ✘ | ✘ | ✘ | ✘ |
| VS | ✔ | ✘ | ✔ | ✘ |
| Ba | ✘ | ✘ | ✘ | ✘ |
| SrTiO | ✔ | ✔ | ✔ | ✔ |
| Al | ✔ | ✔ | ✘ | ✘ |
| GaBN | ✔ | ✔ | ✘ | ✘ |
| AcMnO | ✔ | ✔ | ✔ | ✔ |
| PaTlO | ✔ | ✔ | ✔ | ✔ |
| CdCuN | ✔ | ✘ | ✘ | ✘ |
| HoHSe | ✔ | ✘ | ✔ | ✘ |
| Li | ✘ | ✘ | ✘ | ✘ |
| Cd | ✘ | ✘ | ✘ | ✘ |
| AlCrFe | ✔ | ✘ | ✘ | ✘ |
| ZnCdPt | ✔ | ✘ | ✘ | ✘ |
| EuAlSi | ✔ | ✘ | ✔ | ✔ |
| Sc | ✔ | ✔ | ✔ | ✔ |
| GaSeCl | ✘ | ✘ | ✘ | ✘ |
| CaAgN | ✔ | ✘ | ✔ | ✘ |
| BaAlGe | ✔ | ✔ | ✔ | ✘ |
| K | ✘ | ✘ | ✘ | ✘ |
| KCrO | ✔ | ✘ | ✘ | ✘ |
| TiZnCu | ✔ | ✔ | ✔ | ✔ |
| Ta | ✔ | ✘ | ✘ | ✘ |
| AgBiSeS | ✔ | ✔ | ✘ | ✘ |
| ZrTaNO | ✔ | ✘ | ✔ | ✘ |
| MnAlCuPd | ✔ | ✘ | ✘ | ✘ |
| CsNaICl | ✔ | ✘ | ✔ | ✘ |
| DyThCN | ✔ | ✘ | ✔ | ✘ |
| Li | ✘ | ✘ | ✘ | ✘ |
| SrWNO | ✔ | ✘ | ✘ | ✘ |
| Sr | ✘ | ✘ | ✘ | ✘ |
| ZrCuSiAs | ✘ | ✘ | ✘ | ✘ |
| NdNiSnH | ✘ | ✘ | ✘ | ✘ |
| MnCoSnRh | ✘ | ✘ | ✘ | ✘ |
| Mg | ✘ | ✘ | ✘ | ✘ |
| AlCr | ✔ | ✔ | ✘ | ✘ |
| Y | ✔ | ✘ | ✘ | ✘ |
| Ba | ✘ | ✘ | ✘ | ✘ |
| Accuracy | Overall: | Overall: | Overall: | Overall: |
| Binary: | Binary: | Binary: | Binary: | |
| Ternary: | Ternary: | Ternary: | Ternary: | |
| Quarternary: | Quarternary: | Quarternary: | Quarternary: | |
We observed that ParetoCSP successfully found the most stable structures of all cubic and hexagonal binary crystals and most tetragonal binary crystals in the benchmark dataset. The three unsuccessful binary crystals that ParetoCSP failed to identify their exact structures are Ga

Figure 2. Sample structure prediction by ParetoCSP. Every ground truth structure is followed by the predicted structure. (A- P) show that the structures of MnAl, ZrSe
Now, we analyze the performance of ParetoCSP in terms of the quantitative performance metrics. As mentioned before, we used a set of seven performance metrics to evaluate the prediction performance of different CSP algorithms. The values of each performance metric for all
Quantitative performance metrics of ParetoCSP with M3GNet for the
| Crystal | ED | SD | CD | HD | FP | ||
| For each metric and each failure case, the values, which are greater than the range of exact predictions, are denoted by bold letters to mark as high values that quantitatively shows their non-optimality. Binary, ternary, and quarternary crystals are separated by single horizontal lines. | |||||||
| TiCo | |||||||
| CrPd | |||||||
| GaNi | |||||||
| ZrSe | |||||||
| MnAl | |||||||
| NiS | |||||||
| TiO | |||||||
| NiCl | |||||||
| AlNi | |||||||
| CuBr | |||||||
| VPt | |||||||
| MnCo | |||||||
| BN | |||||||
| GeMo | |||||||
| Ca | |||||||
| Ga | |||||||
| CoAs | |||||||
| Li | |||||||
| VS | |||||||
| Ba | |||||||
| SrTiO | |||||||
| Al | |||||||
| GaBN | |||||||
| AcMnO | |||||||
| PaTlO | |||||||
| CdCuN | |||||||
| HoHSe | |||||||
| Li | |||||||
| Cd | |||||||
| AlCrFe | |||||||
| ZnCdPt | |||||||
| EuAlSi | |||||||
| Sc | |||||||
| GaSeCl | |||||||
| CaAgN | |||||||
| BaAlGe | |||||||
| K | |||||||
| KCrO | |||||||
| TiZnCu | |||||||
| Ta | |||||||
| AgBiSeS | |||||||
| ZrTaNO | |||||||
| MnAlCuPd | |||||||
| CsNaICl | |||||||
| DyThCN | |||||||
| Li | |||||||
| SrWNO | |||||||
| Sr | |||||||
| ZrCuSiAs | |||||||
| NdNiSnH | |||||||
| MnCoSnRh | |||||||
| Mg | |||||||
| AlCr4GaC2 | |||||||
| Y | |||||||
| Ba | |||||||
Performance comparison with GN-OA
As reported in ref[36], the GN-OA algorithm achieved the highest performance when utilizing BO[69] as the optimization algorithm and the MEGNet neural network model as the formation energy predictor to guide the optimization process (default GN-OA). Based on the data presented in Table 2, we observed that GN-OA showed a significantly lower success rate than that of ParetoCSP. In comparison to ParetoCSP, GN-OA achieved an accuracy of only
To understand the deteriorated performance of GN-OA in our benchmark study, firstly, we found that the CSP experiments conducted in the original study of GN-OA[36] primarily focused on small binary crystals, particularly those with a
Figure 3 shows a performance metric value comparison for some sample crystals. For better visualization, we limited the
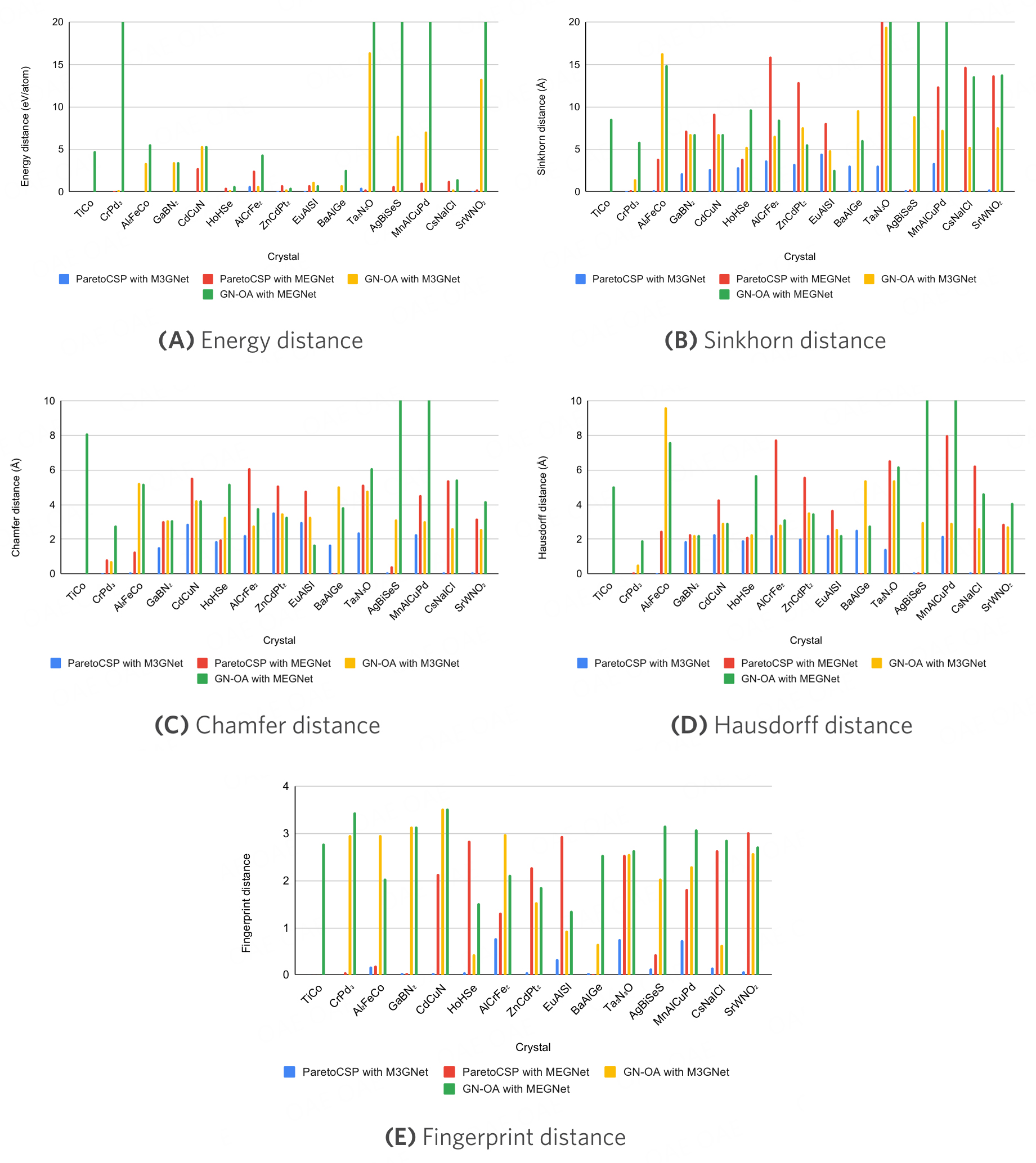
Figure 3. Performance metric comparison of different CSP algorithms evaluated over the sample benchmark crystals. The metric values of ParetoCSP with M3GNet are much smaller (better) than those of other baseline algorithms, which quantitatively shows its superiority. In most cases, GN-OA with MEGNet's metric values is the highest (worst) which is aligned with the observation that it demonstrated the poorest performance among all CSP algorithms.
As discussed in the previous section, M3GNet universal IAP proved to be a better energy predictor than MEGNet. To fairly and objectively evaluate and compare our algorithm's performance, we replaced ParetoCSP's final energy calculator (M3GNet) with the MEGNet GNN for formation energy evaluation. Subsequently, we also replace MEGNet with M3GNet in GN-OA to show that the M3GNet IAP performs better than MEGNet for predicting the most stable energy for CSP. As a result, we ran experiments on four algorithms - ParetoCSP with M3GNet (default ParetoCSP), ParetoCSP with MEGNet, GN-OA with MEGNet (default GN-OA), and GN-OA with M3GNet.
The results of ParetoCSP with M3GNet have been discussed in detail in Section 3.2. ParetoCSP with MEGNet outperformed the default GN-OA by a factor of
Performance comparison of CSP algorithms with different energy models
As discussed in the previous section, M3GNet universal IAP proved to be a better energy predictor than MEGNet. To fairly and objectively evaluate and compare our algorithm's performance, we replaced ParetoCSP's final energy calculator (M3GNet) with the MEGNet GNN for formation energy evaluation. Subsequently, we also replace MEGNet with M3GNet in GN-OA to show that the M3GNet IAP performs better than MEGNet for predicting the most stable energy for CSP. As a result, we ran experiments on four algorithms - ParetoCSP with M3GNet (default ParetoCSP), ParetoCSP with MEGNet, GN-OA with MEGNet (default GN-OA), and GN-OA with M3GNet.
The results of ParetoCSP with M3GNet have been discussed in detail in Section 3.2. ParetoCSP with MEGNet outperformed the default GN-OA by a factor of
Based on the analysis conducted so far, two hypotheses were formulated: firstly, that GN-OA with M3GNet would outperform the default GN-OA, and secondly, that ParetoCSP with M3GNet would outperform GN-OA with M3GNet. As anticipated, GN-OA with M3GNet outperformed the default GN-OA (by a factor of
Parametric study of ParetoCSP
As a multi-objective GA, several hyper-parameters are set before running our ParetoCSP algorithm for CSP. Here, we conducted experiments with our ParetoCSP algorithm with different parameter settings to evaluate their effect. We selected eight crystals for this study containing both successful and unsuccessful predictions, namely TiCo, Ba
Performance results with different hyper-parameters of ParetoCSP with M3GNet
| TiCo | Ba | HoHSe | Cd | SrTiO | GaBN | MnAlCuPd | AgBiSeS | |
| Pop, CP, MP, and Gen denote population size, crossover probability, mutation probability, and total number of generations, respectively. The best results are achieved for a population size of | ||||||||
| Pop | ✘ | ✘ | ✘ | ✘ | ✔ | ✘ | ✘ | ✘ |
| Pop | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✔ |
| Pop | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✔ |
| Pop | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✔ |
| Pop | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ |
| CP | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✘ |
| CP | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✔ |
| CP | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✔ |
| CP | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✘ | ✔ |
| CP | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✔ |
| MP | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✘ |
| MP | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✘ | ✘ |
| MP | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✔ |
| MP | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✘ | ✘ |
| MP | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✘ | ✘ |
| Gen | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ | ✔ | ✘ |
| Gen | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✘ |
| Gen | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✔ |
| Gen | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✔ |
| Gen | ✔ | ✘ | ✔ | ✘ | ✔ | ✔ | ✔ | ✔ |
First, we examined the effect of different population sizes on the selected crystals. We ran the experiments with five different population sizes. The results in Table 4 show that our algorithm performed best with a population size of
Second, we analyzed the performance of our algorithm with varying crossover probabilities. The results indicated that the best performance was achieved with a probability of
Next, we evaluated ParetoCSP's performance with different mutation probabilities and observed that ParetoCSP performed best with a mutation probability of
Finally, we ran experiments with different generations to investigate the impact on algorithm performance. In ref[36], all experiments were run for
Failure case study
ParetoCSP successfully predicted the structures for

Figure 4. Performance metric comparison of structure prediction of different algorithms for the 14 failure cases of ParetoCSP with M3GNet. Despite being inaccurate, ParetoCSP with M3GNet generated structures closer to the corresponding ground truth structures than any other algorithms.
The comparison results for energy distance metric (ED) are presented in Supplementary Figure 2A. We limited the
The observation that Li
Structures of the failed predictions are presented in Supplementary Figure 7. Taking a closer look at the predicted structures, we can clearly notice that, with the exception of some crystals, all of the predictions have the wrong crystal system. Moreover, none of the predicted crystals have the same space group as their corresponding ground truth space group. Furthermore, all the predicted structures deviate significantly from any unstable phase of the same crystal which indicates that the trajectories of the predicted structures by our algorithm did not lead to the right track to find the ground truth crystal, and methods to detect these wrong trajectories in advance needs to be added for better prediction quality.
Trajectory study
To further understand why ParetoCSP works better than GN-OA algorithm, we utilized the multi-dimensional performance metrics of CSP [47] to examine the search patterns of both optimization algorithms employed in ParetoCSP and GN-OA. For most of the crystals, the number of valid structures generated by ParetoCSP is enormous. For better visualization, we selected six crystals for this study which had a comparatively smaller number of valid structures: SrTiO

Figure 5. Trajectories of the traversed structure during the search of different CSP algorithms. (A-C) show the trajectory for SrTiO
Another way to understand the structure search efficiency of ParetoCSP and GN-OA is to check the number of valid structures during the search process. ParetoCSP generated
DISCUSSION
We present ParetoCSP, a CSP algorithm that combines an AFPO enhanced multi-objective GA as an effective structure search function and M3GNet universal IAP as a constructive final energy predictor to achieve efficient structure search for CSP. The objective is to effectively capture the complex relationships between atomic configurations and their corresponding energies. Firstly, ParetoCSP uses the age of a population as a separate optimization criterion. This leads the algorithm to treat the age as a separate dimension in the multi-objective Pareto front where the GA aims to generate structures to minimize the final energy per atom, as well as having low genotypic age. The finding of ref[59] provides a more extensive search process which enables the NSGA-III to perform better as shown in the trajectory results in Section 3.7, where we see that ParetoCSP generated a lot more valid structures during the search process than other evaluated CSP algorithms. This demonstrates the effective exploration of the crystal structure space by ParetoCSP and the efficient identification of the most stable structures.
Overall, we found that ParetoCSP remarkably outperforms the GN-OA algorithm by a factor of
First, we found that for both ParetoCSP and GN-OA, the search process tends to generate a majority of invalid structures even though ParetoCSP works much better than GN-OA. These invalid structures are a waste of search time. Better algorithms that consider crystal symmetry or data-driven generative models may be developed to improve the percentage of valid structures and increase the search efficiency during the search process. In ParetoCSP, the M3GNet IAP is used as the final energy predictor during the search process and structure relaxer after finishing the search process. Compared to MEGNet, M3GNet IAP is proven to be a better choice since after replacing GN-OA's MEGNet with M3GNet IAP, its performance can be improved by a factor of
For the first time, we have used a set of seven quantitative performance metrics to compare and investigate algorithm performances of ParetoCSP and the baselines. We can see from Table 3 that each of the unsuccessful predictions had at least one of the performance metrics value larger than the ground truth value. Additionally, Figure 3 shows that ParetoCSP with M3GNet generated better solutions than any other baseline CSP algorithms as they had much lower performance metric distances (errors) than others. Furthermore, the performance metrics also show that even though ParetoCSP was unable to predict
Inspired by the great success of AlphaFold2 [45] for protein structure prediction, which does not rely on first-principles calculations, we believe that data-driven CSP algorithms based on ML deep neural network energy models have big potential and can reach the same level as AlphaFold2. For this reason, we have focused on the performance comparison with the SOTA GN-OA, a ML potential-based CSP algorithm, and we did not compare our results with CALYPSO [15] and USPEX [14], despite the fact that USPEX also utilizes evolutionary algorithms similar to ours. These algorithms are extremely slow and are not scalable to complex crystals as they depend on ab initio energy calculations, which are computationally very expensive and slow. Currently, they can only deal with simple chemical systems or relatively small crystals (
CONCLUSION
We have introduced an innovative CSP algorithm named ParetoCSP, which synergizes two key components: the multi-objective GA employing AFPO and the M3GNet IAP, for predicting the most stable crystalline material structures. The AFPO-based GA effectively functions as a structure search algorithm, complemented by the role of M3GNet IAP as an efficient final energy predictor that guides the search process. Through comprehensive experimentation involving
DECLARATIONS
Authors' contributions
Conceptualization, resources, supervision, funding acquisition: Hu J
Methodology, writing - original draft preparation: Omee SS, Hu J, Wei L
Software: Omee SS, Hu J
Writing - review and editing: Omee SS, Hu J, Wei L, Hu M
Visualization: Omee SS
Availability of data and materials
The data that support the findings of this study are openly downloadable from the Materials Project database at https://www.materialsproject.org. The source code of our work is freely accessible at https://github.com/sadmanomee/ParetoCSP.
Financial support and sponsorship
The research reported in this work was supported in part by National Science Foundation under the grants 2110033, OAC-2311203, and 2320292. The views, perspectives, and content do not necessarily represent the official views of the NSF.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Oganov AR, Glass CW. Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J Chem Phys 2006;124:244704.
2. Oganov AR, Pickard CJ, Zhu Q, Needs RJ. Structure prediction drives materials discovery. Nat Rev Mater 2019;4:331-48.
3. Hu J, Yang W, Dong R, et al. Contact map based crystal structure prediction using global optimization. CrystEngComm 2021;23:1765-76.
4. Yin X, Gounaris CE. Search methods for inorganic materials crystal structure prediction. Curr Opin Chem Eng 2022;35:100726.
5. Brown N, Ertl P, Lewis R, Luksch T, Reker D, Schneider N. Artificial intelligence in chemistry and drug design. J Comput Aided Mol Des 2020;34:709-15.
6. Kvashnin AG, Allahyari Z, Oganov AR. Computational discovery of hard and superhard materials. J Appl Phys 2019;126:040901.
7. Noh J, Kim J, Stein HS, et al. Inverse design of solid-state materials via a continuous representation. Matter 2019;1:1370-84.
8. Kim B, Lee S, Kim J. Inverse design of porous materials using artificial neural networks. Sci Adv 2020;6:eaax9324.
9. Dan Y, Zhao Y, Li X, Li S, Hu M, Hu J. Generative adversarial networks (GAN) based efficient sampling of chemical composition space for inverse design of inorganic materials. npj Comput Mater 2020;6:84.
10. Long T, Fortunato NM, Opahle I, et al. Constrained crystals deep convolutional generative adversarial network for the inverse design of crystal structures. npj Comput Mater 2021;7:66.
11. Oviedo F, Ren Z, Sun S, et al. Fast and interpretable classification of small X-ray diffraction datasets using data augmentation and deep neural networks. npj Comput Mater 2019;5:60.
12. Pickard CJ, Needs RJ. Ab initio random structure searching. J Phys Condens Matter 2011;23:053201.
13. Woodley SM, Catlow R. Crystal structure prediction from first principles. Nat Mater 2008;7:937-46.
14. Glass CW, Oganov AR, Hansen N. USPEX - evolutionary crystal structure prediction. Comput Phys Commun 2006;175:713-20.
15. Wang Y, Lv J, Zhu L, Ma Y. CALYPSO: a method for crystal structure prediction. Comput Phys Commun 2012;183:2063-70.
17. Sham LJ, Kohn W. One-particle properties of an inhomogeneous interacting electron gas. Phys Rev 1966;145:561-7.
18. Xie T, Grossman JC. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys Rev Lett 2018;120:145301.
19. Chen C, Ye W, Zuo Y, Zheng C, Ong SP. Graph networks as a universal machine learning framework for molecules and crystals. Chem Mater 2019;31:3564-72.
20. Schütt KT, Sauceda HE, Kindermans PJ, Tkatchenko A, Müller KR. SchNet - a deep learning architecture for molecules and materials. J Chem Phys 2018;148:241722.
21. Park CW, Wolverton C. Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery. Phys Rev Mater 2020;4:063801.
22. Omee SS, Louis SY, Fu N, et al. Scalable deeper graph neural networks for high-performance materials property prediction. Patterns 2022;3:100491.
23. Choudhary K, Decost B. Atomistic Line Graph Neural Network for improved materials property predictions. npj Comput Mater 2021;7:185.
24. Kirkpatrick S, Gelatt CD Jr, Vecchi MP. Optimization by simulated annealing. Science 1983;220:671-80.
25. Pannetier J, Bassas-Alsina J, Rodriguez-Carvajal J, Caignaert V. Prediction of crystal structures fromcrystal chemistry rules by simulated annealing. Nature 1990;346:343-5.
26. Schön JC, Jansen M. First step towards planning of syntheses in solid‐state chemistry: determination of promising structure candidates by global optimization. Angew Chem Int Ed Engl 1996;35:1286-304.
28. Martoňák R, Laio A, Bernasconi M, et al. Simulation of structural phase transitions by metadynamics. Z für Krist Cryst Mater 2009;458:182-5.
29. Wales DJ, Doye JPK. Global optimization by basin-hopping and the lowest energy structures of lennard-jones clusters containing up to 110 atoms. J Phys Chem A 1997;101:5111-6.
30. Wales DJ, Scheraga HA. Global optimization of clusters, crystals, and biomolecules. Science 1999;285:1368-72.
31. Goedecker S. Minima hopping: an efficient search method for the global minimum of the potential energy surface of complex molecular systems. J Chem Phys 2004;120:9911-7.
32. Smith RW. Energy minimization in binary alloy models via genetic algorithms. Phys Commun 1992;71:134-46.
33. Woodley SM, Battle PD, Gale JD, Catlow CRA. The prediction of inorganic crystal structures using a genetic algorithm and energy minimisation. Phys Chem Chem Phys 1999;1:2535-42.
34. Lyakhov AO, Oganov AR, Stokes HT, Zhu Q. New developments in evolutionary structure prediction algorithm USPEX. Comput Phys Commun 2013;184:1172-82.
35. Yamashita T, Sato N, Kino H, Miyake T, Tsuda K, Oguchi T. Crystal structure prediction accelerated by Bayesian optimization. Phys Rev Mater 2018;2:013803.
36. Cheng G, Gong XG, Yin WJ. Crystal structure prediction by combining graph network and optimization algorithm. Nat Commun 2022;13:1492.
37. Ryan K, Lengyel J, Shatruk M. Crystal structure prediction via deep learning. J Am Chem Soc 2018;140:10158-68.
38. Hu J, Zhao Y, Li Q, et al. Deep learning-based prediction of contact maps and crystal structures of inorganic materials. ACS Omega 2023;8:26170-9.
39. Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 1996;6:15-50.
40. Soler JM, Artacho E, Gale JD, et al. The SIESTA method for ab initio order- N materials simulation. J Phys Condens Matter 2002;14:2745.
41. Hautier G, Fischer C, Ehrlacher V, Jain A, Ceder G. Data mined ionic substitutions for the discovery of new compounds. Inorg Chem 2011;50:656-63.
42. Wei L, Fu N, Siriwardane EMD, et al. TCSP: a template-based crystal structure prediction algorithm for materials discovery. Inorg Chem 2022;61:8431-9.
43. Kusaba M, Liu C, Yoshida R. Crystal structure prediction with machine learning-based element substitution. Comput Mater Sci 2022;211:111496.
44. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature 2020;577:706-10.
45. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596:583-9.
46. Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021;373:871-6.
47. Wei L, Li Q, Omee SS, Hu J. Towards quantitative evaluation of crystal structure prediction performance. Comput Mater Sci 2024;235:112802.
48. Podryabinkin EV, Tikhonov EV, Shapeev AV, Oganov AR. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning. Phys Rev B 2019;99:064114.
49. Tong Q, Xue L, Lv J, Wang Y, Ma Y. Accelerating CALYPSO structure prediction by data-driven learning of a potential energy surface. Faraday Discuss 2018;211:31-43.
50. Takamoto S, Izumi S, Li J. TeaNet: Universal neural network interatomic potential inspired by iterative electronic relaxations. Comput Mater Sci 2022;207:111280.
51. Takamoto S, Shinagawa C, Motoki D, et al. Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements. Nat Commun 2022;13:2991.
52. Choudhary K, Decost B, Major L, Butler K, Thiyagalingam J, Tavazza F. Unified graph neural network force-field for the periodic table: solid state applications. Digit Discov 2023;2:346-55.
53. Chen C, Ong SP. A universal graph deep learning interatomic potential for the periodic table. Nat Comput Sci 2022;2:718-28.
54. Deng B, Zhong P, Jun K, et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat Mach Intell 2023;5:1031-41.
55. Shao X, Lv J, Liu P, et al. A symmetry-orientated divide-and-conquer method for crystal structure prediction. J Chem Phys 2022;156:014105.
56. Hahn T, Shmueli U, Arthur JCW. International tables for crystallography. 1983. Available from: https://it.iucr.org/. [Last accessed on 22 Feb 2024].
57. Deb K, Jain H. An evolutionary many-objective optimization algorithm using reference-point-based nondominated sorting approach, part Ⅰ: solving problems with box constraints. IEEE Trans Evol Computat 2014;18:577-601.
58. Jain H, Deb K. An evolutionary many-objective optimization algorithm using reference-point based nondominated sorting approach, part Ⅱ: handling constraints and extending to an adaptive approach. IEEE Trans Evol Computat 2014;18:602-22.
59. Schmidt MD, Lipson H. Age-fitness pareto optimization. In: Proceedings of the 12th Annual Conference on Genetic and Evolutionary Computation. New York, NY, USA: Association for Computing Machinery; 2010. pp. 543-4.
60. Isayev O, Oses C, Toher C, Gossett E, Curtarolo S, Tropsha A. Universal fragment descriptors for predicting properties of inorganic crystals. Nat Commun 2017;8:15679.
61. Cheng J, Zhang C, Dong L. A geometric-information-enhanced crystal graph network for predicting properties of materials. Commun Mater 2021;2:92.
62. Yan K, Liu Y, Lin Y, Ji S. Periodic graph transformers for crystal material property prediction. arXiv. [Preprint. ] Sep 23, 2022[accessed 2024 Feb 22]. Available from: https://arxiv.org/abs/2209.11807.
63. Hu J, Goodman E, Seo K, Fan Z, Rosenberg R. The hierarchical fair competition (HFC) framework for sustainable evolutionary algorithms. Evol Comput 2005;13:241-77.
64. Hornby GS. ALPS: the age-layered population structure for reducing the problem of premature convergence. In: Proceedings of the 8th Annual Conference on Genetic and Evolutionary Computation. New York, NY, USA: Association for Computing Machinery; 2006; pp. 815-22. 2006.
65. Schmidt M, Lipson H. Age-fitness pareto optimization. In: Riolo R, Mcconaghy T, Vladislavleva E, editors. Genetic programming theory and practice VⅢ. New York, NY, USA: Springer; 2011. pp. 129-46.
66. Deb K, Pratap A, Agarwal S, Meyarivan T. A fast and elitist multiobjective genetic algorithm: Nsga-Ⅱ. IEEE Trans Evol Comput 2002;6:182-97.
67. Blank J, Deb K, Roy PC. Investigating the normalization procedure of NSGA-Ⅲ. Available from: https://www.egr.msu.edu/~kdeb/papers/c2018009.pdf. [Last accessed on 25 Mar 2024]
68. Jain A, Ong SP, Hautier G, et al. Commentary: The materials project: a materials genome approach to accelerating materials innovation. APL Mater 2013;1:011002.
69. Bergstra J, Yamins D, Cox D. Making a science of model search: hyperparameter optimization in hundreds of dimensions for vision architectures. In: Proceedings of the 30th International Conference on Machine Learning. Atlanta, Georgia, USA: PMLR; 2013. pp. 115-23. Available from: https://proceedings.mlr.press/v28/bergstra13.html. [Last accessed on 22 Feb 2024].
70. van der Maaten L, Hinton G. Visualizing data using t-SNE. 2008. Available from: https://www.jmlr.org/papers/volume9/vandermaaten08a/vandermaaten08a.pdf. [Last accessed on 22 Feb 2024].
Cite This Article

How to Cite
Omee, S. S.; Wei, L.; Hu, M.; Hu, J. Crystal structure prediction using neural network potential and age-fitness Pareto genetic algorithm. J. Mater. Inf. 2024, 4, 2. http://dx.doi.org/10.20517/jmi.2023.33
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.