
Cellular Epigenetic Targets and Epidrugs in Breast Cancer Therapy: Mechanisms, Challenges, and Future Perspectives

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Inclusion and Exclusion Criteria
2.3. Data Extraction and Synthesis
2.4. Quality Assessment
3. Breast Cancer and Epigenetic Regulation
4. Epigenetic Alterations in Breast Cancer
4.1. DNA Methylation in Neoplastic Cells
4.2. DNA Methylation Pattern of the Breast Cancer Microenvironment
4.3. The Role of Non-Coding RNA
4.4. Alterations in Histone Modification
4.5. Mechanism of Epigenetics Related to Estrogen
4.6. Epigenetic Alterations During the EMT
5. Epigenetics and Cancer Progression
6. Drug Resistance and Epigenetics
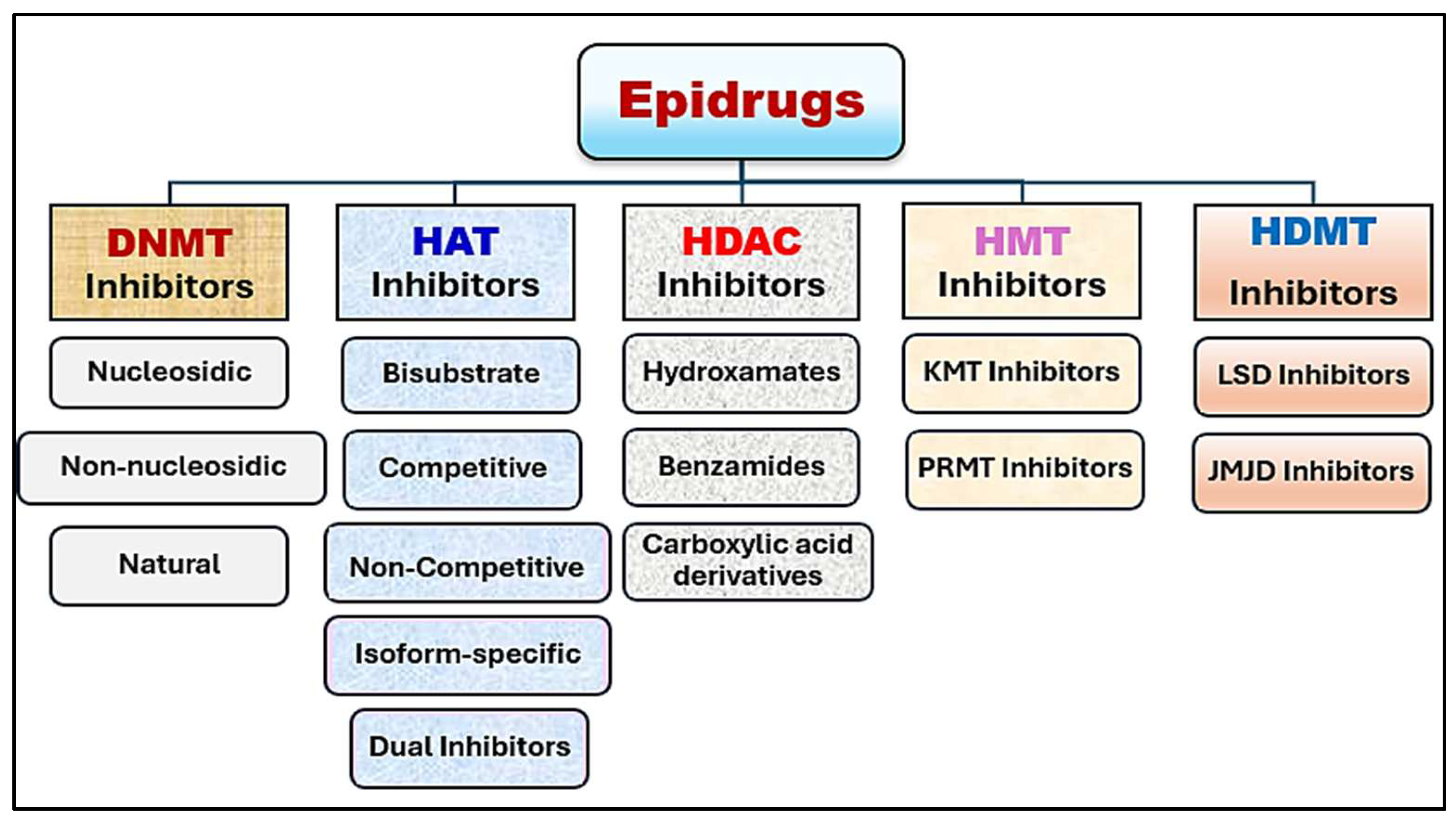
7. Progress in Epigenetic Therapy
7.1. DNA-Modifying Drugs
7.2. HAT and HDAC Inhibitors
7.3. HMT and HDMT Inhibitors
7.4. Epidrug Combination Therapy in Breast Cancer
7.5. RNA-Based Therapies
8. Obstacles and Constraints
8.1. Insufficient Selectivity
8.2. Resistance Emergence
8.3. High Cost
9. Future Directions and Opportunities
9.1. Personalized Epigenetic Therapies
9.2. Combination Therapies
9.3. Targeting Epigenetic Plasticity
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, P.; Aggarwal, R. An Overview of Triple-Negative Breast Cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.-Q.; Sun, H.; Xie, Y.; Patel, H.; Bo, L.; Lin, H.; Chen, Z.-S. Therapeutic Evolution in HR+/HER2-Breast Cancer: From Targeted Therapy to Endocrine Therapy. Front. Pharmacol. 2024, 15, 1340764. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Bertaut, A.; Ladoire, S.; Desmoulins, I.; Jankowski, C.; Beltjens, F.; Charon-Barra, C.; Bergeron, A.; Richard, C.; Boidot, R. HER2-Low Luminal Breast Carcinoma Is Not a Homogenous Clinicopathological and Molecular Entity. Cancers 2024, 16, 2009. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Yalamarty, S.S.K.; Rajmalani, B.A.; Filipczak, N.; Torchilin, V.P. Recent Strategies to Overcome Breast Cancer Resistance. Crit. Rev. Oncol. Hematol. 2024, 197, 104351. [Google Scholar] [CrossRef] [PubMed]
- Golmohammadi, M.; Zamanian, M.Y.; Al-Ani, A.M.; Jabbar, T.L.; Kareem, A.K.; Aghaei, Z.H.; Tahernia, H.; Hjazi, A.; Jissir, S.A.; Hakimizadeh, E. Targeting STAT3 Signaling Pathway by Curcumin and Its Analogues for Breast Cancer: A Narrative Review. Anim. Models Exp. Med. 2024, 7, 853–867. [Google Scholar] [CrossRef]
- Borri, F.; Granaglia, A. Pathology of Triple Negative Breast Cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef]
- So, J.Y.; Ohm, J.; Lipkowitz, S.; Yang, L. Triple Negative Breast Cancer (TNBC): Non-Genetic Tumor Heterogeneity and Immune Microenvironment: Emerging Treatment Options. Pharmacol. Ther. 2022, 237, 108253. [Google Scholar] [CrossRef]
- Varzaru, V.B.; Vlad, T.; Popescu, R.; Vlad, C.S.; Moatar, A.E.; Cobec, I.M. Triple-Negative Breast Cancer: Molecular Particularities Still a Challenge. Diagnostics 2024, 14, 1875. [Google Scholar] [CrossRef]
- Mir, M.A.; Mir, A.Y. Current Treatment Approaches to Breast Cancer. In Therapeutic Potential of Cell Cycle Kinases in Breast Cancer; Mir, M., Ed.; Springer Nature: Singapore, 2023; pp. 23–51. ISBN 978-981-19-8910-0. [Google Scholar]
- Zaami, S.; Melcarne, R.; Patrone, R.; Gullo, G.; Negro, F.; Napoletano, G.; Monti, M.; Aceti, V.; Panarese, A.; Borcea, M.C. Oncofertility and Reproductive Counseling in Patients with Breast Cancer: A Retrospective Study. J. Clin. Med. 2022, 11, 1311. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, Y.; Zhou, X.; Yu, H.; Tan, Y.; Du, Y.; Zhang, Q.; Wu, Y. Current Landscape of Personalized Clinical Treatments for Triple-Negative Breast Cancer. Front. Pharmacol. 2022, 13, 977660. [Google Scholar] [CrossRef]
- Subhan, M.A.; Parveen, F.; Shah, H.; Yalamarty, S.S.K.; Ataide, J.A.; Torchilin, V.P. Recent Advances with Precision Medicine Treatment for Breast Cancer Including Triple-Negative Sub-Type. Cancers 2023, 15, 2204. [Google Scholar] [CrossRef] [PubMed]
- Obidiro, O.; Battogtokh, G.; Akala, E.O. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics 2023, 15, 1796. [Google Scholar] [CrossRef] [PubMed]
- Chang-Qing, Y.; Jie, L.; Shi-Qi, Z.; Kun, Z.; Zi-Qian, G.; Ran, X.; Hui-Meng, L.; Ren-Bin, Z.; Gang, Z.; Da-Chuan, Y. Recent Treatment Progress of Triple Negative Breast Cancer. Prog. Biophys. Mol. Biol. 2020, 151, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chatterjee, M.; Ghosh, P.; Ganguly, K.K.; Basu, M.; Ghosh, M.K. Targeting PD-1/PD-L1 in Cancer Immunotherapy: An Effective Strategy for Treatment of Triple-Negative Breast Cancer (TNBC) Patients. Genes Dis. 2023, 10, 1318–1350. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, C.; Tao, Y.; Lin, W.; Wang, P. Immunotherapy for Triple-Negative Breast Cancer. Pharmaceutics 2021, 13, 2003. [Google Scholar] [CrossRef]
- Syrnioti, A.; Petousis, S.; Newman, L.A.; Margioula-Siarkou, C.; Papamitsou, T.; Dinas, K.; Koletsa, T. Triple Negative Breast Cancer: Molecular Subtype-Specific Immune Landscapes with Therapeutic Implications. Cancers 2024, 16, 2094. [Google Scholar] [CrossRef]
- Wang, J.; Li, L.; Xu, Z.P. Enhancing Cancer Chemo-immunotherapy: Innovative Approaches for Overcoming Immunosuppression by Functional Nanomaterials. Small Methods 2024, 8, 2301005. [Google Scholar] [CrossRef]
- Galluzzi, L.; Garg, A. Immune Checkpoint Biology in Health and Disease; Elsevier: Amsterdam, The Netherlands, 2024; Volume 382, ISBN 0-443-13574-6. [Google Scholar]
- Prabhu, K.S.; Sadida, H.Q.; Kuttikrishnan, S.; Junejo, K.; Bhat, A.A.; Uddin, S. Beyond Genetics: Exploring the Role of Epigenetic Alterations in Breast Cancer. Pathol.-Res. Pract. 2024, 254, 155174. [Google Scholar] [CrossRef]
- Guo, L.; Kong, D.; Liu, J.; Zhan, L.; Luo, L.; Zheng, W.; Zheng, Q.; Chen, C.; Sun, S. Breast Cancer Heterogeneity and Its Implication in Personalized Precision Therapy. Exp. Hematol. Oncol. 2023, 12, 3. [Google Scholar] [CrossRef]
- Madnoorkar, N.; Jain, R.; Sapate, R.; Bhamra, G.S.; Thakare, P. Drug Resistance in Breast Cancer. OncoReview 2024, 14, 23–33. [Google Scholar] [CrossRef]
- Zhou, L.; Yu, C.-W. Epigenetic Modulations in Triple-Negative Breast Cancer: Therapeutic Implications for Tumor Microenvironment. Pharmacol. Res. 2024, 204, 107205. [Google Scholar] [CrossRef] [PubMed]
- Abiola, S.A.; Ben-Chioma, A.E.; Fidelis, B.G.; Aloy, S.C.; Elekima, I. Epigenetic Modulation in Breast Cancer: From Mechanisms to Therapeutic Interventions. Int. Res. J. Oncol. 2024, 7, 1–13. [Google Scholar]
- Yin, J.; Gu, T.; Chaudhry, N.; Davidson, N.E.; Huang, Y. Epigenetic Modulation of Antitumor Immunity and Immunotherapy Response in Breast Cancer: Biological Mechanisms and Clinical Implications. Front. Immunol. 2024, 14, 1325615. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.; Domingos, C.; Figueiredo, A.; Hamilton, R.; Tabori, U.; Castelo-Branco, P. Cancer Stem Cells in Prostate Cancer: Implications for Targeted Therapy. Urol. Int. 2017, 99, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Yin, D.; Wang, Y.; Du, W.; Qin, Y.; Ding, A.; Li, H. Cancer Stem Cells and Combination Therapies to Eradicate Them. Curr. Pharm. Des. 2020, 26, 1994–2008. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Florean, C.; Dicato, M.; Diederich, M. Epigenetic Alterations as a Universal Feature of Cancer Hallmarks and a Promising Target for Personalized Treatments. Curr. Top. Med. Chem. 2016, 16, 745–776. [Google Scholar] [CrossRef]
- Jin, M.C.; Connolly, I.D.; Ravi, K.; Tobert, D.G.; MacDonald, S.M.; Shin, J.H. Unraveling Molecular Advancements in Chordoma Tumorigenesis and Treatment Response: A Review of Scientific Discoveries and Clinical Implications. Neurosurg. Focus 2024, 56, E18. [Google Scholar] [CrossRef]
- Bayramova, J.; Tarakci, E.; Huseynova, G.; Yazici, H.; Yazici, H. Genetic Alterations in Lung Cancer. Turk. J. Oncol. 2024, 39, 234–243. [Google Scholar]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic Heterogeneity in Cancer. Biomark. Res. 2019, 7, 1–19. [Google Scholar] [CrossRef]
- Temian, D.C.; Pop, L.A.; Irimie, A.I.; Berindan-Neagoe, I. The Epigenetics of Triple-Negative and Basal-like Breast Cancer: Current Knowledge. J. Breast Cancer 2018, 21, 233–243. [Google Scholar] [CrossRef]
- Sarvari, P.; Sarvari, P.; Ramírez-Díaz, I.; Mahjoubi, F.; Rubio, K. Advances of Epigenetic Biomarkers and Epigenome Editing for Early Diagnosis in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 9521. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Yan, Q. The Roles of Epigenetics in Cancer Progression and Metastasis. Biochem. J. 2021, 478, 3373–3393. [Google Scholar] [CrossRef] [PubMed]
- Salarinia, R.; Sahebkar, A.; Peyvandi, M.; Reza Mirzaei, H.; Reza Jaafari, M.; Matbou Riahi, M.; Ebrahimnejad, H.; Sadri Nahand, J.; Hadjati, J.; Ostadi Asrami, M. Epi-Drugs and Epi-miRs: Moving beyond Current Cancer Therapies. Curr. Cancer Drug Targets 2016, 16, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Venkatesh, D.; Kandasamy, T.; Ghosh, S.S. Epigenetic Modulations in Breast Cancer: An Emerging Paradigm in Therapeutic Implications. Front. Biosci.-Landmark 2024, 29, 287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Hashimoto, H.; Horton, J.R.; Zhang, X. Mechanisms of DNA Methylation, Methyl-CpG Recognition, and Demethylation in Mammals. In Handbook of Epigenetics; Elsevier: San Diego, CA, USA, 2011; pp. 9–24. [Google Scholar]
- Liu, R.; Zhao, E.; Yu, H.; Yuan, C.; Abbas, M.N.; Cui, H. Methylation across the Central Dogma in Health and Diseases: New Therapeutic Strategies. Signal Transduct. Target. Ther. 2023, 8, 310. [Google Scholar]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Zhu, D.; Zeng, S.; Su, C.; Li, J.; Xuan, Y.; Lin, Y.; Xu, E.; Fan, Q. The Interaction between DNA Methylation and Tumor Immune Microenvironment: From the Laboratory to Clinical Applications. Clin. Epigenetics 2024, 16, 24. [Google Scholar] [CrossRef]
- Hazra, A.; Bose, P.; Sunita, P.; Pattanayak, S.P. Molecular Epigenetic Dynamics in Breast Carcinogenesis. Arch. Pharm. Res. 2021, 44, 741–763. [Google Scholar] [CrossRef]
- de Ruijter, T.C.; van der Heide, F.; Smits, K.M.; Aarts, M.J.; van Engeland, M.; Heijnen, V.C. Prognostic DNA Methylation Markers for Hormone Receptor Breast Cancer: A Systematic Review. Breast Cancer Res. 2020, 22, 1–12. [Google Scholar] [CrossRef]
- Ali, R.W.; Jassim, T.S. The Role of Methylation in Gene Expression and Complex Human Diseases. Int. J. Appl. Chem. Biol. Sci. 2023, 4, 37–61. [Google Scholar]
- Khan, M.A. Epigenetics Involvement in Breast Cancer. In Breast Cancer: From Bench to Personalized Medicine; Springer: Berlin/Heidelberg, Germany, 2022; pp. 145–183. [Google Scholar]
- Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Córdoba, A.; de Azúa, Y.R.; Aguiar, B.; Beloqui, R.; Armendáriz, P.; Arriola, M.; Martín-Sánchez, E. ADAM12 Is a Potential Therapeutic Target Regulated by Hypomethylation in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 903. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zheng, X.; Ren, L.; Fu, W.; Liu, J.; Xv, J.; Liu, S.; Wang, J.; Du, G. Epigenetic Hypomethylation and Upregulation of GD3s in Triple Negative Breast Cancer. Ann. Transl. Med. 2019, 7, 723. [Google Scholar] [CrossRef] [PubMed]
- Kasprowicz, A.; Sophie, G.-D.; Lagadec, C.; Delannoy, P. Role of GD3 Synthase ST8Sia I in Cancers. Cancers 2022, 14, 1299. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, X.; Pang, X.; Li, L.; Wang, J.; Yang, C.; Du, G. Ganglioside GD3 Synthase (GD3S), a Novel Cancer Drug Target. Acta Pharm. Sin. B 2018, 8, 713–720. [Google Scholar] [CrossRef]
- Wan, H.; Li, Z.; Wang, H.; Cai, F.; Wang, L. ST8SIA1 Inhibition Sensitizes Triple Negative Breast Cancer to Chemotherapy via Suppressing Wnt/β-Catenin and FAK/Akt/mTOR. Clin. Transl. Oncol. 2021, 23, 902–910. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Wong, W.K.; Yin, B.; Lam, C.Y.K.; Huang, Y.; Yan, J.; Tan, Z.; Wong, S.H.D. The Interplay between Epigenetic Regulation and CD8+ T Cell Differentiation/Exhaustion for T Cell Immunotherapy. Front. Cell Dev. Biol. 2022, 9, 783227. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization by DNA Methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic Modulation of Antitumor Immunity for Improved Cancer Immunotherapy. Mol. Cancer 2021, 20, 1–27. [Google Scholar] [CrossRef]
- Wei, J.-W.; Huang, K.; Yang, C.; Kang, C.-S. Non-Coding RNAs as Regulators in Epigenetics. Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Esteller, M. Non-Coding RNAs, Epigenetics, and Cancer: Tying It All Together. Cancer Metastasis Rev. 2018, 37, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Zhang, M.; Xu, Y.; Wang, J.; Ding, N.; Yu, Y.; Sun, S.; Dai, W.; Xue, X.; Li, N.; et al. Non-Coding RNAs as Therapeutic Targets in Cancer and Its Clinical Application. J. Pharm. Anal. 2024, 14, 100947. [Google Scholar] [CrossRef] [PubMed]
- Beňačka, R.; Szabóová, D.; Guľašová, Z.; Hertelyová, Z.; Radoňak, J. Non-Coding RNAs in Human Cancer and Other Diseases: Overview of the Diagnostic Potential. Int. J. Mol. Sci. 2023, 24, 16213. [Google Scholar] [CrossRef]
- Khalighfard, S.; Alizadeh, A.M.; Irani, S.; Omranipour, R. Plasma miR-21, miR-155, miR-10b, and Let-7a as the Potential Biomarkers for the Monitoring of Breast Cancer Patients. Sci. Rep. 2018, 8, 17981. [Google Scholar] [CrossRef]
- Luo, Q.; Li, X.; Gao, Y.; Long, Y.; Chen, L.; Huang, Y.; Fang, L. MiRNA-497 Regulates Cell Growth and Invasion by Targeting Cyclin E1 in Breast Cancer. Cancer Cell Int. 2013, 13, 1–8. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef]
- Kaur, J.; Salehen, N.; Norazit, A.; Rahman, A.A.; Murad, N.A.A.; Rahman, N.M.A.N.A.; Ibrahim, K. Tumor Suppressive Effects of GAS5 in Cancer Cells. Non-Coding RNA 2022, 8, 39. [Google Scholar] [CrossRef]
- Filippova, E.A.; Fridman, M.V.; Burdennyy, A.M.; Loginov, V.I.; Pronina, I.V.; Lukina, S.S.; Dmitriev, A.A.; Braga, E.A. Long Noncoding RNA GAS5 in Breast Cancer: Epigenetic Mechanisms and Biological Functions. Int. J. Mol. Sci. 2021, 22, 6810. [Google Scholar] [CrossRef]
- Acharjee, S.; Chauhan, S.; Pal, R.; Tomar, R.S. Mechanisms of DNA Methylation and Histone Modifications. Prog. Mol. Biol. Transl. Sci. 2023, 197, 51–92. [Google Scholar]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 1596. [Google Scholar] [CrossRef]
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li, H. Histone Modifications and Their Role in Colorectal Cancer. Pathol. Oncol. Res. 2020, 26, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, S.A.; Sahu, K.K.; Sengupta, S.; Partap, S.; Karpoormath, R.; Kumar, B.; Kumar, D. Recent Advancement of HDAC Inhibitors against Breast Cancer. Med. Oncol. 2023, 40, 201. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Visvader, J.E. Dysregulation of Histone Methyltransferases in Breast Cancer—Opportunities for New Targeted Therapies? Mol. Oncol. 2016, 10, 1497–1515. [Google Scholar] [CrossRef] [PubMed]
- Adibfar, S.; Elveny, M.; Kashikova, H.S.; Mikhailova, M.V.; Farhangnia, P.; Vakili-Samiani, S.; Tarokhian, H.; Jadidi-Niaragh, F. The Molecular Mechanisms and Therapeutic Potential of EZH2 in Breast Cancer. Life Sci. 2021, 286, 120047. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, H.; Luo, Y.; Tong, S.; Liu, Y. EZH2: The Roles in Targeted Therapy and Mechanisms of Resistance in Breast Cancer. Biomed. Pharmacother. 2024, 175, 116624. [Google Scholar] [CrossRef]
- Kurani, H.; Razavipour, S.F.; Harikumar, K.B.; Dunworth, M.; Ewald, A.J.; Nasir, A.; Pearson, G.; Van Booven, D.; Zhou, Z.; Azzam, D. DOT1L Is a Novel Cancer Stem Cell Target for Triple-Negative Breast Cancer. Clin. Cancer Res. 2022, 28, 1948–1965. [Google Scholar] [CrossRef]
- Tong, D.; Tang, Y.; Zhong, P. The Emerging Roles of Histone Demethylases in Cancers. Cancer Metastasis Rev. 2024, 43, 795–821. [Google Scholar] [CrossRef]
- Pei, J.; Zhang, S.; Yang, X.; Han, C.; Pan, Y.; Li, J.; Wang, Z.; Sun, C.; Zhang, J. Epigenetic Regulator KDM4A Activates Notch1-NICD-Dependent Signaling to Drive Tumorigenesis and Metastasis in Breast Cancer. Transl. Oncol. 2023, 28, 101615. [Google Scholar] [CrossRef]
- Gaughan, L.; Stockley, J.; Coffey, K.; O’Neill, D.; Jones, D.L.; Wade, M.; Wright, J.; Moore, M.; Tse, S.; Rogerson, L. KDM4B Is a Master Regulator of the Estrogen Receptor Signalling Cascade. Nucleic Acids Res. 2013, 41, 6892–6904. [Google Scholar] [CrossRef]
- Wu, X.; Deng, Y.; Zu, Y.; Yin, J. Histone Demethylase KDM4C Activates HIF1α/VEGFA Signaling through the Costimulatory Factor STAT3 in NSCLC. Am. J. Cancer Res. 2020, 10, 491. [Google Scholar]
- Noirrit-Esclassan, E.; Valera, M.-C.; Tremollieres, F.; Arnal, J.-F.; Lenfant, F.; Fontaine, C.; Vinel, A. Critical Role of Estrogens on Bone Homeostasis in Both Male and Female: From Physiology to Medical Implications. Int. J. Mol. Sci. 2021, 22, 1568. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, B.; Ou-Yang, L. Role of Estrogen Receptors in Health and Disease. Front. Endocrinol. 2022, 13, 839005. [Google Scholar] [CrossRef] [PubMed]
- Moisand, A.; Madéry, M.; Boyer, T.; Domblides, C.; Blaye, C.; Larmonier, N. Hormone Receptor Signaling and Breast Cancer Resistance to Anti-Tumor Immunity. Int. J. Mol. Sci. 2023, 24, 15048. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Lukina, E.; Friedemann, M.; Menschikowski, M.; Hagelgans, A.; Aliev, G. The Crucial Role of Epigenetic Regulation in Breast Cancer Anti-Estrogen Resistance: Current Findings and Future Perspectives. Semin. Cancer Biol. 2022, 82, 35–59. [Google Scholar] [CrossRef]
- Ayaz, G.; Yasar, P.; Olgun, C.E.; Karakaya, B.; Kars, G.; Razizadeh, N.; Yavuz, K.; Turan, G.; Muyan, M. Dynamic Transcriptional Events Mediated by Estrogen Receptor Alpha. Front. Biosci.-Landmark 2019, 24, 245–276. [Google Scholar] [CrossRef]
- Howard, E.W.; Yang, X. microRNA Regulation in Estrogen Receptor-Positive Breast Cancer and Endocrine Therapy. Biol. Proced. Online 2018, 20, 1–19. [Google Scholar] [CrossRef]
- Adams, B.D.; Furneaux, H.; White, B.A. The Micro-Ribonucleic Acid (miRNA) miR-206 Targets the Human Estrogen Receptor-α (ERα) and Represses ERα Messenger RNA and Protein Expression in Breast Cancer Cell Lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef]
- Song, J.; Ouyang, Y.; Che, J.; Li, X.; Zhao, Y.; Yang, K.; Zhao, X.; Chen, Y.; Fan, C.; Yuan, W. Potential Value of miR-221/222 as Diagnostic, Prognostic, and Therapeutic Biomarkers for Diseases. Front. Immunol. 2017, 8, 56. [Google Scholar] [CrossRef]
- Song, Q.; An, Q.; Niu, B.; Lu, X.; Zhang, N.; Cao, X. Role of miR-221/222 in Tumor Development and the Underlying Mechanism. J. Oncol. 2019, 2019, 7252013. [Google Scholar] [CrossRef]
- Dudás, J.; Ladányi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism That Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef]
- Nowak, E.; Bednarek, I. Aspects of the Epigenetic Regulation of EMT Related to Cancer Metastasis. Cells 2021, 10, 3435. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Gui, X.; Qu, W.; Zhang, X. Expressions and Clinical Significance of CCN5 and E-Cadherin in Primary and Recurrent Lesions of Breast Cancer. Front. Genet. 2024, 15, 1404515. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-P.; Kuang, J.-Y.; Yang, Q.-K.; Bian, X.-W.; Yu, S.-C. Beyond a Tumor Suppressor: Soluble E-cadherin Promotes the Progression of Cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef]
- Park, M.; Kang, K.W.; Kim, J.W. The Role and Application of Transcriptional Repressors in Cancer Treatment. Arch. Pharm. Res. 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Rahimian, A.; Barati, G.; Mehrandish, R.; Mellati, A.A. Inhibition of Histone Deacetylases Reverses Epithelial-Mesenchymal Transition in Triple-Negative Breast Cancer Cells through a Slug Mediated Mechanism. Mol. Biol. 2018, 52, 406–413. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, W.; Liu, S.; Chen, C. Targeting Breast Cancer Stem Cells. Int. J. Biol. Sci. 2023, 19, 552. [Google Scholar] [CrossRef]
- Muñoz, P.; Iliou, M.S.; Esteller, M. Epigenetic Alterations Involved in Cancer Stem Cell Reprogramming. Mol. Oncol. 2012, 6, 620–636. [Google Scholar] [CrossRef]
- Wils, L.J.; Bijlsma, M.F. Epigenetic Regulation of the Hedgehog and Wnt Pathways in Cancer. Crit. Rev. Oncol. Hematol. 2018, 121, 23–44. [Google Scholar] [CrossRef]
- Klarmann, G.J.; Decker, A.; Farrar, W.L. Epigenetic Gene Silencing in the Wnt Pathway in Breast Cancer. Epigenetics 2008, 3, 59–63. [Google Scholar] [CrossRef]
- Suzuki, H.; Toyota, M.; Caraway, H.; Gabrielson, E.; Ohmura, T.; Fujikane, T.; Nishikawa, N.; Sogabe, Y.; Nojima, M.; Sonoda, T.; et al. Frequent Epigenetic Inactivation of Wnt Antagonist Genes in Breast Cancer. Br. J. Cancer 2008, 98, 1147–1156. [Google Scholar] [CrossRef]
- Sharma, A.; Mir, R.; Galande, S. Epigenetic Regulation of the Wnt/β-Catenin Signaling Pathway in Cancer. Front. Genet. 2021, 12, 681053. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.; Chiang, H.-C.; Kaklamani, V.G. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2–ERα–GREB1 Transcriptional Axis. Cancer Res. 2018, 78, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/ΚDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; McCuaig, R.D.; Tan, A.H.; Tu, W.J.; Wu, F.; Wagstaff, K.M.; Zafar, A.; Ali, S.; Diwakar, H.; Dahlstrom, J.E. Selective Targeting of Protein Kinase C (PKC)-θ Nuclear Translocation Reduces Mesenchymal Gene Signatures and Reinvigorates Dysfunctional CD8+ T Cells in Immunotherapy-Resistant and Metastatic Cancers. Cancers 2022, 14, 1596. [Google Scholar] [CrossRef]
- Ghosh, A.; Himaja, A.; Biswas, S.; Kulkarni, O.; Ghosh, B. Advances in the Delivery and Development of Epigenetic Therapeutics for the Treatment of Cancer. Mol. Pharm. 2023, 20, 5981–6009. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone Deacetylases and Their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Montalvo-Casimiro, M.; González-Barrios, R.; Meraz-Rodriguez, M.A.; Juárez-González, V.T.; Arriaga-Canon, C.; Herrera, L.A. Epidrug Repurposing: Discovering New Faces of Old Acquaintances in Cancer Therapy. Front. Oncol. 2020, 10, 605386. [Google Scholar] [CrossRef]
- Yang, T.; Yang, Y.; Wang, Y. Predictive Biomarkers and Potential Drug Combinations of Epi-Drugs in Cancer Therapy. Clin. Epigenetics 2021, 13, 113. [Google Scholar] [CrossRef]
- Kim, A.; Mo, K.; Kwon, H.; Choe, S.; Park, M.; Kwak, W.; Yoon, H. Epigenetic Regulation in Breast Cancer: Insights on Epidrugs. Epigenomes 2023, 7, 6. [Google Scholar] [CrossRef]
- Davletgildeeva, A.T.; Kuznetsov, N.A. The Role of DNMT Methyltransferases and TET Dioxygenases in the Maintenance of the DNA Methylation Level. Biomolecules 2024, 14, 1117. [Google Scholar] [CrossRef]
- Mehdipour, P.; Murphy, T.; De Carvalho, D.D. The Role of DNA-Demethylating Agents in Cancer Therapy. Pharmacol. Ther. 2020, 205, 107416. [Google Scholar] [CrossRef] [PubMed]
- Akone, S.H.; Ntie-Kang, F.; Stuhldreier, F.; Ewonkem, M.B.; Noah, A.M.; Mouelle, S.E.M.; Müller, R. Natural Products Impacting DNA Methyltransferases and Histone Deacetylases. Front. Pharmacol. 2020, 11, 992. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Magaña, M.J.; Martínez-Aguilar, R.; Lucendo, E.; Campillo-Davo, D.; Schulze-Osthoff, K.; Ruiz-Ruiz, C. The Antihypertensive Drug Hydralazine Activates the Intrinsic Pathway of Apoptosis and Causes DNA Damage in Leukemic T Cells. Oncotarget 2016, 7, 21875. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Gonzalez, A.; Coronel, J.; Cetina, L.; González-Fierro, A.; Chavez-Blanco, A.; Taja-Chayeb, L. Hydralazine–Valproate: A Repositioned Drug Combination for the Epigenetic Therapy of Cancer. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1433–1444. [Google Scholar] [CrossRef]
- Billam, M.; Sobolewski, M.D.; Davidson, N.E. Effects of a Novel DNA Methyltransferase Inhibitor Zebularine on Human Breast Cancer Cells. Breast Cancer Res. Treat. 2010, 120, 581–592. [Google Scholar] [CrossRef]
- Altundag, O.; Altundag, K.; Gunduz, M. DNA Methylation Inhibitor, Procainamide, May Decrease the Tamoxifen Resistance by Inducing Overexpression of the Estrogen Receptor Beta in Breast Cancer Patients. Med. Hypotheses 2004, 63, 684–687. [Google Scholar] [CrossRef]
- Asl, M.M.; Asl, J.M.; Naghitorabi, M. Comparison of the Effects of Olsalazine and Decitabine on the Expression of CDH1 and uPA Genes and Cytotoxicity in MDA-MB-231 Breast Cancer Cells. Res. Pharm. Sci. 2021, 16, 278–285. [Google Scholar] [CrossRef]
- Chequin, A.; Costa, L.E.; de Campos, F.F.; Moncada, A.D.; de Lima, L.T.; Sledz, L.R.; Picheth, G.F.; Adami, E.R.; Acco, A.; Goncalves, M.B. Antitumoral Activity of Liraglutide, a New DNMT Inhibitor in Breast Cancer Cells in Vitro and in Vivo. Chem. Biol. Interact. 2021, 349, 109641. [Google Scholar] [CrossRef]
- Alkaff, A.H.; Saragih, M.; Imana, S.N.; Nasution, M.A.F.; Tambunan, U.S.F. Identification of DNA Methyltransferase-1 Inhibitor for Breast Cancer Therapy through Computational Fragment-Based Drug Design. Molecules 2021, 26, 375. [Google Scholar] [CrossRef]
- Demetriadou, C.; Koufaris, C.; Kirmizis, A. Histone N-Alpha Terminal Modifications: Genome Regulation at the Tip of the Tail. Epigenetics Chromatin 2020, 13, 29. [Google Scholar] [CrossRef]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef] [PubMed]
- Alsamri, H.; Hasasna, H.E.; Baby, B.; Alneyadi, A.; Dhaheri, Y.A.; Ayoub, M.A.; Eid, A.H.; Vijayan, R.; Iratni, R. Carnosol Is a Novel Inhibitor of p300 Acetyltransferase in Breast Cancer. Front. Oncol. 2021, 11, 664403. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.M.; Abdelghany, M.K.; Messmer, M.; Yue, B.; Deeves, S.E.; Kindle, K.B.; Mantelingu, K.; Aslam, A.; Winkler, G.S.; Kundu, T.K. Differential Effects of Garcinol and Curcumin on Histone and P53 Modifications in Tumour Cells. BMC Cancer 2013, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent Developments of HDAC Inhibitors: Emerging Indications and Novel Molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. [Google Scholar] [CrossRef]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Lai, J.-C.; Hou, M.-F.; Lin, C.-S. Combination of Tacedinaline and EHMT2 Inhibition Increases Breast Cancer Cell Death Involving BIRC5 Repression and GADD45A Induction. Cancer Res. 2019, 79, 4715. [Google Scholar] [CrossRef]
- Kaya Çakir, H.; Eroglu, O. In Vitro Anti-Proliferative Effect of Capecitabine (Xeloda) Combined with Mocetinostat (MGCD0103) in 4T1 Breast Cancer Cell Line by Immunoblotting. Iran. J. Basic Med. Sci. 2021, 24, 1515–1522. [Google Scholar] [CrossRef]
- Shi, M.-Q.; Xu, Y.; Fu, X.; Pan, D.-S.; Lu, X.-P.; Xiao, Y.; Jiang, Y.-Z. Advances in Targeting Histone Deacetylase for Treatment of Solid Tumors. J. Hematol. Oncol.J Hematol Oncol 2024, 17, 37. [Google Scholar] [CrossRef]
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.J.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- D’Oto, A.; Tian, Q.; Davidoff, A.M.; Yang, J. Histone Demethylases and Their Roles in Cancer Epigenetics. J. Med. Oncol. Ther. 2016, 1, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, T.; Malik, L.; McCuaig, R.D.; Tu, W.J.; Wu, F.; Lim, P.S.; Tan, A.H.; Dahlstrom, J.E.; Clingan, P.; Moylan, E. A Phase 1 Proof of Concept Study Evaluating the Addition of an LSD1 Inhibitor to Nab-Paclitaxel in Advanced or Metastatic Breast Cancer (EPI-PRIMED). Front. Oncol. 2022, 12, 862427. [Google Scholar] [CrossRef]
- Alsaad, I.; Abdel Rahman, D.M.; Al-Tamimi, O.; Alhaj, S.A.; Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K. Targeting MAO-B with Small-Molecule Inhibitors: A Decade of Advances in Anticancer Research (2012–2024). Molecules 2024, 30, 126. [Google Scholar] [CrossRef]
- Satram-Maharaj, T.; Nyarko, J.N.; Kuski, K.; Fehr, K.; Pennington, P.R.; Truitt, L.; Freywald, A.; Lukong, K.E.; Anderson, D.H.; Mousseau, D.D. The Monoamine Oxidase-A Inhibitor Clorgyline Promotes a Mesenchymal-to-Epithelial Transition in the MDA-MB-231 Breast Cancer Cell Line. Cell Signal. 2014, 26, 2621–2632. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, F.; Sun, F. ORY-1001, a KDM1A Inhibitor, Inhibits Proliferation, and Promotes Apoptosis of Triple Negative Breast Cancer Cells by Inactivating Androgen Receptor. Drug Dev. Res. 2022, 83, 208–216. [Google Scholar] [CrossRef]
- Pan, C.-H.; Chang, Y.-F.; Lee, M.-S.; Wen, B.-C.; Ko, J.-C.; Liang, S.-K.; Liang, M.-C. Vorinostat Enhances the Cisplatin-Mediated Anticancer Effects in Small Cell Lung Cancer Cells. BMC Cancer 2016, 16, 1–11. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Roca, M.S.; Zotti, A.I.; Leone, A.; Bruzzese, F.; Vitagliano, C.; Scogliamiglio, G.; Russo, D.; D’Angelo, G.; Franco, R. Valproic Acid Potentiates the Anticancer Activity of Capecitabine in Vitro and in Vivo in Breast Cancer Models via Induction of Thymidine Phosphorylase Expression. Oncotarget 2015, 7, 7715. [Google Scholar] [CrossRef]
- Laengle, J.; Kabiljo, J.; Hunter, L.; Homola, J.; Prodinger, S.; Egger, G.; Bergmann, M. Histone Deacetylase Inhibitors Valproic Acid and Vorinostat Enhance Trastuzumab-Mediated Antibody-Dependent Cell-Mediated Phagocytosis. J. Immunother. Cancer 2020, 8, e000195. [Google Scholar] [CrossRef]
- Buocikova, V.; Longhin, E.M.; Pilalis, E.; Mastrokalou, C.; Miklikova, S.; Cihova, M.; Poturnayova, A.; Mackova, K.; Babelova, A.; Trnkova, L. Decitabine Potentiates Efficacy of Doxorubicin in a Preclinical Trastuzumab-Resistant HER2-Positive Breast Cancer Models. Biomed. Pharmacother. 2022, 147, 112662. [Google Scholar] [CrossRef]
- Ramaswamy, B.; Fiskus, W.; Cohen, B.; Pellegrino, C.; Hershman, D.L.; Chuang, E.; Luu, T.; Somlo, G.; Goetz, M.; Swaby, R. Phase I–II Study of Vorinostat plus Paclitaxel and Bevacizumab in Metastatic Breast Cancer: Evidence for Vorinostat-Induced Tubulin Acetylation and Hsp90 Inhibition in Vivo. Breast Cancer Res. Treat. 2012, 132, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Pattarawat, P.; Wallace, S.; Pfisterer, B.; Odoi, A.; Wang, H.-C.R. Formulation of a Triple Combination Gemcitabine plus Romidepsin+ Cisplatin Regimen to Efficaciously and Safely Control Triple-Negative Breast Cancer Tumor Development. Cancer Chemother. Pharmacol. 2020, 85, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Arce, C.; Perez-Plasencia, C.; Gonzalez-Fierro, A.; de la Cruz-Hernandez, E.; Revilla-Vazquez, A.; Chavez-Blanco, A.; Trejo-Becerril, C.; Perez-Cardenas, E.; Taja-Chayeb, L.; Bargallo, E. A Proof-of-Principle Study of Epigenetic Therapy with Hydralazine and Magnesium Valproate plus Doxorubicin Cyclophosphamide as Neoadjuvant Therapy for Locally Advanced Breast Cancer. BMC Cancer 2007, 7, 1–2. [Google Scholar] [CrossRef]
- Medon, M.; Vidacs, E.; Vervoort, S.J.; Li, J.; Jenkins, M.R.; Ramsbottom, K.M.; Trapani, J.A.; Smyth, M.J.; Darcy, P.K.; Atadja, P.W. HDAC Inhibitor Panobinostat Engages Host Innate Immune Defenses to Promote the Tumoricidal Effects of Trastuzumab in HER2+ Tumors. Cancer Res. 2017, 77, 2594–2606. [Google Scholar] [CrossRef]
- Lee, J.; Bartholomeusz, C.; Mansour, O.; Humphries, J.; Hortobagyi, G.N.; Ordentlich, P.; Ueno, N.T. A Class I Histone Deacetylase Inhibitor, Entinostat, Enhances Lapatinib Efficacy in HER2-Overexpressing Breast Cancer Cells through FOXO3-Mediated Bim1 Expression. Breast Cancer Res. Treat. 2014, 146, 259–272. [Google Scholar] [CrossRef]
- Won, K.-A.; Spruck, C. Triple-negative Breast Cancer Therapy: Current and Future Perspectives. Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase I Study of Panobinostat (LBH589) and Letrozole in Postmenopausal Metastatic Breast Cancer Patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef]
- Connolly, R.M.; Li, H.; Jankowitz, R.C.; Zhang, Z.; Rudek, M.A.; Jeter, S.C.; Slater, S.A.; Powers, P.; Wolff, A.C.; Fetting, J.H. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin. Cancer Res. 2017, 23, 2691–2701. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, T.; Geng, C.; Zhang, Y.; Zhang, J.; Ning, Z.; Jiang, Z. Exploratory Clinical Study of Chidamide, an Oral Subtype-Selective Histone Deacetylase Inhibitor, in Combination with Exemestane in Hormone Receptor-Positive Advanced Breast Cancer. Chin. J. Cancer Res. 2018, 30, 605. [Google Scholar] [CrossRef]
- Vernier, M.; McGuirk, S.; Dufour, C.R.; Wan, L.; Audet-Walsh, E.; St-Pierre, J.; Giguère, V. Inhibition of DNMT1 and ERRα Crosstalk Suppresses Breast Cancer via Derepression of IRF4. Oncogene 2020, 39, 6406–6420. [Google Scholar] [CrossRef]
- Issa, J.-P.; Garcia-Manero, G.; Huang, X.; Cortes, J.; Ravandi, F.; Jabbour, E.; Borthakur, G.; Brandt, M.; Pierce, S.; Kantarjian, H.M. Results of Phase 2 Randomized Study of Low-dose Decitabine with or without Valproic Acid in Patients with Myelodysplastic Syndrome and Acute Myelogenous Leukemia. Cancer 2015, 121, 556–561. [Google Scholar] [CrossRef]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The Role of Histone Deacetylase Inhibitors in Metastatic Breast Cancer. Breast 2019, 43, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Pattarawat, P.; Hunt, J.T.; Poloway, J.; Archibald, C.J.; Wang, H.-C.R. A Triple Combination Gemcitabine+romidepsin+cisplatin to Effectively Control Triple-Negative Breast Cancer Tumor Development, Recurrence, and Metastasis. Cancer Chemother. Pharmacol. 2021, 88, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Prabhakaran, R.; Thamarai, R.; Sivasamy, S.; Dhandayuthapani, S.; Batra, J.; Kamaraj, C.; Karthik, K.; Shah, M.A.; Mallik, S. Epigenetic Frontiers: miRNAs, Long Non-Coding RNAs and Nanomaterials Are Pioneering to Cancer Therapy. Epigenetics Chromatin 2024, 17, 31. [Google Scholar] [CrossRef]
- Alpuche-Lazcano, S.P.; Scarborough, R.J.; Gatignol, A. MicroRNAs and Long Non-Coding RNAs during Transcriptional Regulation and Latency of HIV and HTLV. Retrovirology 2024, 21, 5. [Google Scholar] [CrossRef]
- Stanton, S.E.; Gad, E.; Corulli, L.R.; Lu, H.; Disis, M.L. Tumor-Associated Antigens Identified Early in Mouse Mammary Tumor Development Can Be Effective Vaccine Targets. Vaccine 2019, 37, 3552–3561. [Google Scholar] [CrossRef]
- Sifuentes-Romero, I.; Milton, S.L.; García-Gasca, A. Post-Transcriptional Gene Silencing by RNA Interference in Non-Mammalian Vertebrate Systems: Where Do We Stand? Mutat. Res. Mutat. Res. 2011, 728, 158–171. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and Other Non-Coding RNAs as Targets for Anticancer Drug Development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Pérez-Moreno, P.; Pérez, Y.; Calaf, G.M. The Role of MicroRNAs in Breast Cancer and the Challenges of Their Clinical Application. Diagnostics 2023, 13, 3072. [Google Scholar] [CrossRef]
- Traber, G.M.; Yu, A.-M. RNAi-Based Therapeutics and Novel RNA Bioengineering Technologies. J. Pharmacol. Exp. Ther. 2023, 384, 133–154. [Google Scholar] [CrossRef]
- Slaughter, M.J.; Shanle, E.K.; Khan, A.; Chua, K.F.; Hong, T.; Boxer, L.D.; Allis, C.D.; Josefowicz, S.Z.; Garcia, B.A.; Rothbart, S.B.; et al. HDAC Inhibition Results in Widespread Alteration of the Histone Acetylation Landscape and BRD4 Targeting to Gene Bodies. Cell Rep. 2021, 34, 108638. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. P53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or Not Specific Recruitment of DNMTs for DNA Methylation, an Epigenetic Dilemma. Clin. Epigenetics 2018, 10, 17. [Google Scholar] [CrossRef]
- Minisini, M.; Mascaro, M.; Brancolini, C. HDAC-Driven Mechanisms in Anticancer Resistance: Epigenetics and Beyond. Cancer Drug Resist. 2024, 7, 46. [Google Scholar] [CrossRef]
- Prabhu, K.S.; Kuttikrishnan, S.; Ahmad, N.; Habeeba, U.; Mariyam, Z.; Suleman, M.; Bhat, A.A.; Uddin, S. H2AX: A Key Player in DNA Damage Response and a Promising Target for Cancer Therapy. Biomed. Pharmacother. 2024, 175, 116663. [Google Scholar] [CrossRef]
- Shen, C.; Li, M.; Duan, Y.; Jiang, X.; Hou, X.; Xue, F.; Zhang, Y.; Luo, Y. HDAC Inhibitors Enhance the Anti-Tumor Effect of Immunotherapies in Hepatocellular Carcinoma. Front. Immunol. 2023, 14, 1170207. [Google Scholar] [CrossRef]
- Mungly, S.B.; Peter, E.P.; Hii, L.-W.; Mai, C.-W.; Chung, F.F.-L. Epigenetic Drug Interventions in Breast Cancer: A Narrative Review of Current Research and Future Directions. Prog. Microbes Mol. Biol. 2024, 7, 1–40. [Google Scholar] [CrossRef]
- Passaro, A.; Al Bakir, M.; Hamilton, E.G.; Diehn, M.; André, F.; Roy-Chowdhuri, S.; Mountzios, G.; Wistuba, I.I.; Swanton, C.; Peters, S. Cancer Biomarkers: Emerging Trends and Clinical Implications for Personalized Treatment. Cell 2024, 187, 1617–1635. [Google Scholar] [CrossRef]
- Hexem, E.; Taha, T.A.-E.A.; Dhemesh, Y.; Baqar, M.A.; Nada, A. Deciphering Glioblastoma: Unveiling Imaging Markers for Predicting MGMT Promoter Methylation Status. Curr. Probl. Cancer 2025, 54, 101156. [Google Scholar] [CrossRef]
- Pathak, A.; Tomar, S.; Pathak, S. Epigenetics and Cancer: A Comprehensive Review. Asian Pac. J. Cancer Biol. 2023, 8, 75–89. [Google Scholar] [CrossRef]
- Costa, P.M.d.S.; Sales, S.L.A.; Pinheiro, D.P.; Pontes, L.Q.; Maranhão, S.S.; Pessoa, C.d.Ó.; Furtado, G.P.; Furtado, C.L.M. Epigenetic Reprogramming in Cancer: From Diagnosis to Treatment. Front. Cell Dev. Biol. 2023, 11, 1116805. [Google Scholar] [CrossRef] [PubMed]
- Trnkova, L.; Buocikova, V.; Mego, M.; Cumova, A.; Burikova, M.; Bohac, M.; Miklikova, S.; Cihova, M.; Smolkova, B. Epigenetic Deregulation in Breast Cancer Microenvironment: Implications for Tumor Progression and Therapeutic Strategies. Biomed. Pharmacother. 2024, 174, 116559. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Wang, Y.; Zhang, Y.; Yang, Y.; Cao, J.; Huang, J.; Chen, J.; Chen, H.; Zhang, J.; Wang, L. Leveraging CRISPR Gene Editing Technology to Optimize the Efficacy, Safety and Accessibility of CAR T-Cell Therapy. Leukemia 2024, 38, 2517–2543. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhao, H.; Wang, R.; Chen, Y.; Ouyang, X.; Li, W.; Sun, Y.; Peng, A. Cancer Epigenetics: From Laboratory Studies and Clinical Trials to Precision Medicine. Cell Death Discov. 2024, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Casari, G.; Romaldi, B.; Scirè, A.; Minnelli, C.; Marzioni, D.; Ferretti, G.; Armeni, T. Epigenetic Properties of Compounds Contained in Functional Foods Against Cancer. Biomolecules 2025, 15, 15. [Google Scholar] [CrossRef]
- Tao, L.; Zhou, Y.; Luo, Y.; Qiu, J.; Xiao, Y.; Zou, J.; Zhang, Y.; Liu, X.; Yang, X.; Gou, K.; et al. Epigenetic Regulation in Cancer Therapy: From Mechanisms to Clinical Advances. MedComm-Oncol. 2024, 3, e59. [Google Scholar] [CrossRef]
- Prebet, T.; Sun, Z.; Ketterling, R.P.; Zeidan, A.; Greenberg, P.; Herman, J.; Juckett, M.; Smith, M.R.; Malick, L.; Paietta, E.; et al. Azacitidine with or without Entinostat for the Treatment of Therapy-Related Myeloid Neoplasm: Further Results of the E1905 North American Leukemia Intergroup Study. Br. J. Haematol. 2016, 172, 384–391. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Jen, W.-Y.; Kantarjian, H.; Kadia, T.M.; DiNardo, C.D.; Issa, G.C.; Short, N.J.; Yilmaz, M.; Borthakur, G.; Ravandi, F.; Daver, N.G. Combination Therapy with Novel Agents for Acute Myeloid Leukaemia: Insights into Treatment of a Heterogenous Disease. Br. J. Haematol. 2024, 205, 30–47. [Google Scholar] [CrossRef]
- Foo, J.; Basanta, D.; Rockne, R.C.; Strelez, C.; Shah, C.; Ghaffarian, K.; Mumenthaler, S.M.; Mitchell, K.; Lathia, J.D.; Frankhouser, D.; et al. Roadmap on Plasticity and Epigenetics in Cancer. Phys. Biol. 2022, 19, 031501. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epidrug Generation | Class | Compounds | Reference |
|---|---|---|---|
| First generation | DNMT inhibitors | Azacitidine, decitabine | [101,102] |
| HDAC inhibitors | Trapoxin A, trichostatin, vorinostat, romidepsin | [101,102] | |
| Second generation | DNMT inhibitors | Hydralazine, guadecitabine, CP-4200, zebularine | [102,103] |
| HDAC inhibitors | Panobinostat, belinostat, dacinostat, quisinostat, CUDC-101, entinostat, chidamide, tacedinaline, tefinostat, pivanex, butyric acid, valproic acid, phenylbutyric acid | [102,103] | |
| Third generation | KDM inhibitors | Clorgyline, GSK2816126, bizine, KDM5-C70, ORY-101, JIB-04, 4SC-202, tranylcypromine, pargyline | [102,103] |
| KMT inhibitors | Tazemostat, Pinometostat, Sinefungine, BIX-01294, GSK2816126, GSK3326595, GSK3368715, JNJ64619178, DZNep, CPI360, GSK343, EPZ004777, UNC0638, UNC0224 | [102,103] | |
| Bromodomain ligands | CPI-0610, RVX-280, I-BET762, OTX015 | [102,103] |
| S. No. | Drug Combination | Mechanism of Combined Action | Reference |
|---|---|---|---|
| 1 | Decitabine + zebularine | Disturbance of colony formation potential and cell proliferation | [109] |
| 2 | Exemestane + entinostat | Aromatase inhibitor and HDAC class I inhibitor | [101] |
| 3 | Zebularine + vorinostat | Disturbance of colony formation potential and cell proliferation | [109] |
| 4 | Capecitabine + valproic acid | Increase in TP, decrease in TS enzymes, decrease in thymidine synthesis | [132] |
| 5 | Valproic acid + vorinostat + Trustuzumab | Decrease in MCL1, increase in ADCC and ADCP | [133] |
| 6 | Paclitaxel + liraglutide | Stimulation of cellular demethylation via the abolition of DNMTs and transcription of ADAM33, CDH1, and ESR1 genes, leading to inhibition of cell migration and viability | [112] |
| 7 | Methotrexate + liraglutide | Stimulation of cellular demethylation via the abolition of DNMTs and transcription of ADAM33, CDH1, and ESR1 genes, leading to inhibition of cell migration and viability | [112] |
| 8 | Vorinostat + olaparib | PARP, HDAC | [119] |
| 9 | UNC0638 + tacedinaline | Modulation of G9a and class I HDAC, inhibition of BIRC5, and stimulation of GADD45A | [121] |
| 10 | Doxorubicin + decitabine | Inhibition of tumor proliferation, DNMT1 activity, and DNA methylation | [134] |
| 11 | Paclitaxel + vorinostat | Activation of acetylation of both α-tubulin and histone, proteasomal breakdown of Hsp90, enhancement in antiangiogenetic effect | [135] |
| 12 | Nab-paclitaxel + phenelzine | Inhibition of CSC generation by downregulation of mesenchymal markers | [127] |
| 13 | Gemcitabine + romidepsin + cisplatin | TNBC cell apoptosis via ROS generation | [136] |
| 14 | Doxorubicin + hydralazine + cyclophosphamide + magnesium valproate | DNA demethylation and inhibition of HDAC activity, decrease in C5me content | [137] |
| 15 | Panobinostat + trustuzumab | Stimulation of NK-cell-mediated immune response | [138] |
| 16 | Lapatinib + entinostat | Transcriptional activation of FOXO3 and Bim | [139] |
| 17 | Atezolizumab + entinostat | Downregulation of HDAC activity and PD | [140] |
| 18 | Panobinostat + letrozole | BC cell sensitization to hormonal therapy, stimulation of H3, H4 acetylation, inhibition of aromatase activity | [141] |
| 19 | Entinostat + azacitidine | Inhibition of DNA synthesis and HDAC class I activity | [142] |
| 20 | Chidamide + mocetinostat | Inhibition of aromatase and HDAC subtype activities | [143] |
| 21 | Decitabine + C29 | Inhibition of DNMT1 and ERRα | [144] |
| 22 | Mocetinostat + capecitabine | Stimulation of cell apoptosis pathway by inhibiting HDAC1, BC12, Akt, c-myc, and PI3K and stimulating C-Parp, cas-7, Bax, Cas-9, Pten, Cas-3, and p-53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alalhareth, I.S.; Alyami, S.M.; Alshareef, A.H.; Ajeibi, A.O.; Al Munjem, M.F.; Elfifi, A.A.; Alsharif, M.M.; Alzahrani, S.A.; Alqaad, M.A.; Bakir, M.B.; et al. Cellular Epigenetic Targets and Epidrugs in Breast Cancer Therapy: Mechanisms, Challenges, and Future Perspectives. Pharmaceuticals 2025, 18, 207. https://doi.org/10.3390/ph18020207
Alalhareth IS, Alyami SM, Alshareef AH, Ajeibi AO, Al Munjem MF, Elfifi AA, Alsharif MM, Alzahrani SA, Alqaad MA, Bakir MB, et al. Cellular Epigenetic Targets and Epidrugs in Breast Cancer Therapy: Mechanisms, Challenges, and Future Perspectives. Pharmaceuticals. 2025; 18(2):207. https://doi.org/10.3390/ph18020207
Chicago/Turabian StyleAlalhareth, Ibrahim S., Saleh M. Alyami, Ali H. Alshareef, Ahmed O. Ajeibi, Manea F. Al Munjem, Ahmad A. Elfifi, Meshal M. Alsharif, Seham A. Alzahrani, Mohammed A. Alqaad, Marwa B. Bakir, and et al. 2025. "Cellular Epigenetic Targets and Epidrugs in Breast Cancer Therapy: Mechanisms, Challenges, and Future Perspectives" Pharmaceuticals 18, no. 2: 207. https://doi.org/10.3390/ph18020207
APA StyleAlalhareth, I. S., Alyami, S. M., Alshareef, A. H., Ajeibi, A. O., Al Munjem, M. F., Elfifi, A. A., Alsharif, M. M., Alzahrani, S. A., Alqaad, M. A., Bakir, M. B., & Abdel-Wahab, B. A. (2025). Cellular Epigenetic Targets and Epidrugs in Breast Cancer Therapy: Mechanisms, Challenges, and Future Perspectives. Pharmaceuticals, 18(2), 207. https://doi.org/10.3390/ph18020207