

Abstract
The Arabidopsis genome contains numerous large duplicated chromosomal segments, but the different approaches used in previous analyses led to different interpretations regarding the number and timing of ancestral large-scale duplication events. Here, using more appropriate methodology and a more recent version of the genome sequence annotation, we investigate the scale and timing of segmental duplications in Arabidopsis. We used protein sequence similarity searches to detect duplicated blocks in the genome, used the level of synonymous substitution between duplicated genes to estimate the relative ages of the blocks containing them, and analyzed the degree of overlap between adjacent duplicated blocks. We conclude that the Arabidopsis lineage underwent at least two distinct episodes of duplication. One was a polyploidy that occurred much more recently than estimated previously, before the Arabidopsis/Brassica rapa split and probably during the early emergence of the crucifer family (24–40 Mya). An older set of duplicated blocks was formed after the monocot/dicot divergence, and the relatively low level of overlap among these blocks indicates that at least some of them are remnants of a larger duplication such as a polyploidy or aneuploidy.
The complete genome sequence of Arabidopsis thaliana (Arabidopsis Genome Initiative 2000), a model diploid plant species, provides raw information on all genes and proteins needed during the lifetime of a flowering plant and allows fine analysis of an essential cellular structure, the chromosome. Given the small size of the Arabidopsis genome, one of the most surprising discoveries revealed by its sequence is that, like the Saccharomyces cerevisiae (Wolfe and Shields 1997) and human (Lander et al. 2001; Venter et al. 2001; Gu et al. 2002; McLysaght et al. 2002) genomes, it contains numerous large duplicated chromosomal segments (Arabidopsis Genome Initiative 2000; Blanc et al. 2000; Paterson et al. 2000; Vision et al. 2000).
Although the Arabidopsis genome has clearly been shaped by large-scale DNA duplications, the number, extent, and timing of these duplications have been controversial (Sankoff 2001; Wolfe 2001). On the one hand, each pair of sister regions may come from an independent duplication of a chromosome segment. On the other hand, several pairs of sister regions may originate from a single large-scale duplication (i.e., aneuploidy or polyploidy), later followed by genomic rearrangements that split up and relocate the original duplicated sequences around the genome, a process referred to as diploidization (Wolfe 2001). Two key features can be used to distinguish between these two scenarios. Firstly, because different pairs of sister regions originating from a single large-scale duplication event must have been formed simultaneously, molecular clock analysis is expected to show the same time of divergence. Secondly, in contrast to random and independent segmental duplications in which the regions involved can overlap each other by chance, a single large-scale duplication followed by chromosomal rearrangements is expected not to show any overlap between adjacent duplicated regions.
The first systematic studies of block duplications in the Arabidopsis genome used nucleotide alignment and revealed that most of the genome is found in duplicate (Arabidopsis Genome Initiative 2000; Blanc et al. 2000; Paterson et al. 2000). Because the identified duplicated regions did not overlap each other, it was hypothesized that an Arabidopsis ancestor underwent one polyploidy event (Arabidopsis Genome Initiative 2000; Blanc et al. 2000). However, Vision et al. (2000), using a sensitive approach based on protein–protein alignments, reported a level of segmental duplications that was much greater than observed previously. Importantly, many duplicated regions identified in their study were overlapping, which is indicative of multiple events at different times. Using a molecular clock approach, they divided the 103 pairs of regions into 4 different classes of coalescent age and concluded that 4 different large-scale duplication events occurred in the Arabidopsis lineage. However, a weakness of the Vision et al. (2000) analysis is that their dating method was based on the assumption that different groups of proteins have the same median rate of evolution. Because there is significant heterogeneity in the rate of amino acid substitutions among different proteins, this approach is questionable (Sankoff 2001; Wolfe 2001) and may have led to erroneous interpretations about both the number of events and their dates. This prompted us to reinvestigate the number and timing of duplications in Arabidopsis. We confirm that a complete polyploidy occurred, but estimate that it occurred much more recently than proposed previously. We also find evidence for a second, much older polyploidy that has been partly obscured by other segmental duplications.
RESULTS
Block Detection
We searched the genome of Arabidopsis for pairs of duplicated regions using protein sequence similarity to define gene paralogy. Sister regions are defined as two chromosome segments sharing similar genes in the same order, and further characterized by the number of duplicated gene pairs linking them (termed sm; McLysaght et al. 2002). This approach detected 108 pairs of blocks sharing 6 or more duplicated genes (sm ≥ 6; Fig. 1). The statistical significance of these pairs of regions was assessed by re-running the block detection algorithm on 1000 different shuffled gene maps. The mean number of block pairs with sm ≥ 6 found in randomized genomes is <1 (mean 0.24, SD 0.71), suggesting that all 108 blocks depicted on the map likely result from duplication of chromosomal regions. Moreover, it should be pointed out that even for blocks of smaller size (sm < 6), the number of block pairs in the real genome is very significantly higher than what would be expected by chance, indicating that other genuine duplicated chromosomal regions of smaller size exist. Blocks of all sizes can be viewed interactively at http://wolfe.gen.tcd.ie/athal/dup.
Figure 1.

Map of duplicated regions identified in the Arabidopsis genome. Red and blue colored boxes depict recent and old sister regions, respectively, as defined by the analysis of the level of synonymous substitutions between duplicated genes. Lines link paired blocks. Centromeres are shown as circles. An interactive version of this map is available at http://wolfe.gen.tcd.ie/athal/dup.
Overall, the block pairs of sm ≥ 6 cover 71% of the genome, with 39% of the length of blocks overlapping other blocks (Fig. 1). They involve 5826 distinct duplicated genes (23% of the proteome), with the largest containing 283 duplicated genes. These results are similar to those of Vision et al. (2000), reflecting the similarities between the two approaches for this part of the analysis.
It is noteworthy that no duplicated blocks were detected within centromeric or pericentromeric regions. Although transcribed genes are known to reside in these parts of the genome (Arabidopsis Genome Initiative 2000), the regions are very enriched with repetitive sequences, decreasing the number of links between possible sister regions and leaving them difficult to detect with our approach.
Level of Synonymous Substitutions Between Duplicated Genes
To assess the relative chronology of duplication events, we estimated the level of synonymous substitutions (Ks) between duplicated genes. Nucleotide substitutions in protein-coding sequences either result in amino acid change (nonsynonymous substitutions) or do not (synonymous substitutions). Because natural selection acts mainly on protein sequences, synonymous codon positions in Arabidopsis are probably largely free from selection, and so accumulate changes in a neutral manner, at a rate similar to the mutation rate. This aspect of our approach differs markedly from that of Vision et al. (2000) who dated duplication events assuming that the average rate of nonsynonymous substitution was the same for all groups of duplicated genes.
For each pair of sister regions, we obtained the distribution of Ks values estimated from each pair of duplicated genes, excluding all values of Ks > 10, because those sequences are highly saturated at synonymous sites and therefore uninformative. Because all of the genes forming a duplicated block were almost certainly duplicated simultaneously (regardless of the actual duplication mechanism), their Ks values can be regarded as an independent random sample derived from genes duplicated at the same time. We therefore used the median Ks value between sister regions to compare their relative duplication dates. The duplicated blocks fall into two major age groups as indicated by Ks levels (Fig. 2). The first group is characterized by 45 block pairs with relatively low Ks. Their median Ks values range from 0.72 to 0.99, suggesting a relatively young age. These regions will be referred to as “recent”, below. Interestingly, their median Ks values are consistent with the conspicuous secondary peak centered on Ks = 0.8 in the distribution obtained by Lynch and Conery (2000), who concluded that this reflected a genome duplication event.
Figure 2.

Box plots of Ks values (Yang 1999) estimated from duplicated genes located in sister regions. The central box depicts the middle 50% of the data between the 25th and 75th percentile for each pair of duplicated blocks, and the enclosed horizontal line represents the median value of the distribution. Asterisks outside of the main bodies of data indicate extreme values.
In contrast, the second group corresponds to 63 block pairs with much greater Ks values. Their medians range from 1.82–6.03, indicating earlier origins (referred to as “old” blocks, below). The higher Ks variance and fewer data points for this class of block pairs did not allow us to subdivide it reliably into further groups, contrary to the recent results of Simillion et al. (2002), who reported two distinct old age classes.
As well as differing in age, these two groups of sister regions also differ markedly from each other in structure. The recent blocks (median, 700 kb; range, 69–4632 kb) are generally more than twice as long as the old ones (median, 284 kb; range, 90–1178 kb). The content of duplicated genes is higher in recent blocks (mean density 28.0% ± 7.8% duplicated genes) than in old ones (mean density 13.5% ± 5.0% duplicated genes). These observations suggest that the older a duplicated region is, the shorter it is, and the less it shares duplicated genes with its partner. This is understandable if we assume progressive gene loss and chromosomal rearrangements over time. This shows, as observed previously in the Arabidopsis genome (Arabidopsis Genome Initiative 2000; Blanc et al. 2000; Paterson et al. 2000; Vision et al. 2000) and in wheat neopolyploids (Kashkush et al. 2002), that a large fraction of the originally duplicated genes returned to a single copy state.
The recent block pairs (shown in red in Fig. 1) cover at least 70% of the genome (80 Mb), or 80% if centromeric regions are not considered. Remarkably, the recent blocks fit perfectly together with essentially no overlap. The cumulative overlap among recent blocks totals only 0.15% of their length, which probably only reflects the incorrect definition of a few duplicated genes at the ends of blocks. The absence of overlap among the recent blocks (Fig. 1), despite their extensive coverage of the genome, together with their homogeneous age (Fig. 2), indicates very clearly that the recent blocks are the remains of a polyploidy event that was followed by gene loss and chromosomal rearrangements (Arabidopsis Genome Initiative 2000; Blanc et al. 2000; Paterson et al. 2000; Bevan et al. 2001; Gu and Huang 2002).
The genomic organization of old duplicated blocks, shown in blue in Figure 1, does not immediately reveal any clear evidence of additional large-scale duplication events. Instead, they only partially cover the genome (33.2 Mb, 26%) and show a substantial degree of overlap (cumulative overlap 25%), suggesting multiple origins. This is explored further below.
Reconstruction of the Ancestral Gene Order
Old duplicated blocks may be difficult to discover for several reasons. Because recent blocks cover the majority of the Arabidopsis genome, most of the old blocks are likely to overlap with them completely. However, after the recent polyploidy, chromosomal rearrangements coupled with large numbers of gene deletions (making ∼70% of loci single copy again) led to the breakage of ancestral gene order and redistribution of genes across new pairs of sister regions. Thus, the recent polyploidy substantially rearranged the genome organization, and could make it difficult to find any sister regions that existed before that event. Applying looser criteria for block detection (allowing larger gaps or considering smaller sized blocks) would probably reveal additional old duplicated blocks, but at the cost of increasing false positives.
We therefore used an alternative approach; we reconstructed the approximate gene order of the ancestral genome that existed prior to the recent polyploidy event, and then searched for old duplicated blocks in the reconstructed genome. Assuming that all genes occurring in recently duplicated blocks resided in the same ancestral region before duplication, the approximate gene order of the ancestral region can be reconstructed by merging genes lying in both sister regions. Using a pseudo genome in which all the recently duplicated blocks have been merged should make it easier to detect older duplication events, because this would emulate the matching pattern of the ancestral genome. Thus, for each pair of recent sister regions, we used the order of the duplicated genes as a framework and filled the intervals between them by alternately interleaving the single copy genes located in the equivalent gaps in the two regions. We constructed the pseudo ancestral genome by taking the real genome as reference, walking from the top of chromosome 1 to the bottom of chromosome 5. Each time a recent block was met, it was interleaved with its corresponding sister (retaining only the longest copy of each duplicated gene) and added to the pseudo genome. Genes located in the sister region were subsequently removed from the real genome in such a way that a merged block appears only once in the pseudo genome. Genes located in unduplicated regions of the real genome were included in the pseudo genome without changing their locations.
The resulting pseudo ancestral genome contained 20,187 genes arranged in a linear array (an ordered list of gene names in the reconstructed genome is available at wolfe.gen.tcd.ie/athal/dup). This was used as a template for the block detection program, which found 68 block pairs with sm ≥ 7 (Fig. 3). These block pairs have a high statistical significance, because experiments in which gene order in the pseudo genome was randomized gave, on average, less than one block pair of sm ≥ 7 (mean 0.68; SD 0.81, among 1000 replicates; we used sm ≥ 6 as a cutoff for block size in the real genome, but sm ≥ 7 in the reconstructed genome, because in each case, this is the size at which less than one block is expected by chance). They cover a larger portion of the reconstructed ancestral genome (49%) than the significant old blocks identified in the real genome (26%), with an average of 13.3% ± 4.0% of their genes duplicated. As observed in the real genome, they show a significant degree of overlap (cumulative overlap 21%). Analysis of the Ks distributions within these blocks did not allow any subdivision into distinct age classes (data not shown), mainly because of the large variance of Ks among blocks, but confirmed their relatively ancient appearance (Ks medians ranging from 1.24–7.25).
Figure 3.
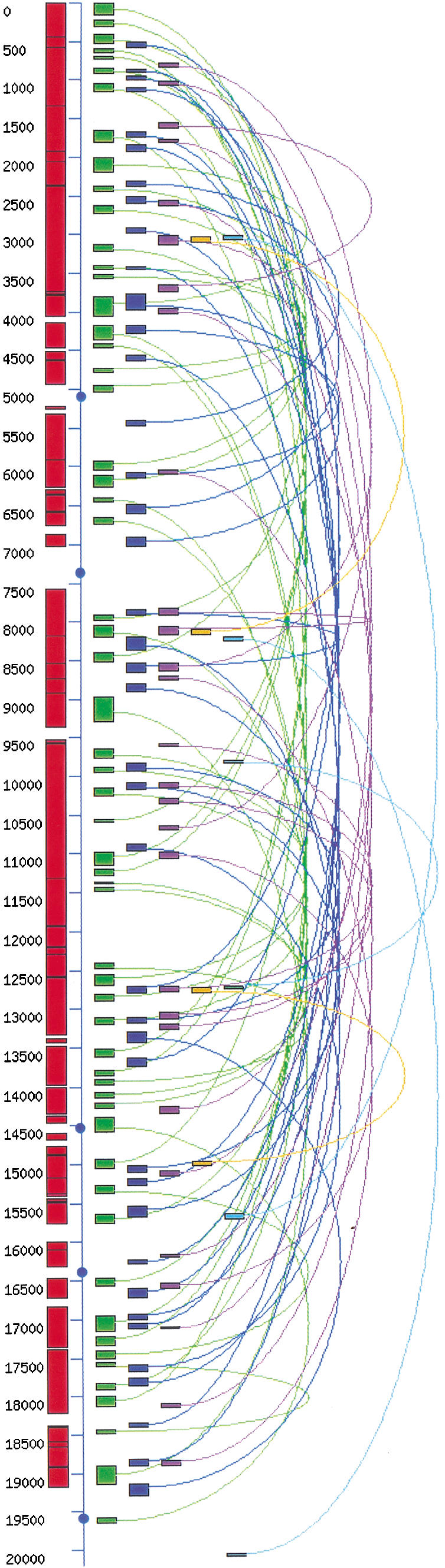
Map of duplicated regions identified in the Arabidopsis pseudo ancestral genome. The vertical line represents a single chromosome containing 20,187 genes ordered according to a procedure that merged the recent blocks (see text) to approximate the ancestral Arabidopsis genome before the last polyploidy event. Red boxes depict the sections of the pseudo genome corresponding to merged sister regions. Other shaded boxes at right represent duplicated regions identified in the pseudo ancestral genome (colors are used for clarity and do not depict any age classification). Curved lines link paired regions, and circles depict the regions corresponding to the centromeres in the real genome.
Analysis of the Degree of Overlap
Although adjacent duplicated blocks detected in the pseudo ancestral genome overlap markedly, indicating multiple origins, their presence could result from large-scale duplication events, or random and independent duplications of chromosomal segments, or a combination of both. To investigate the possibility that large-scale duplication events (such as polyploidy) might have formed some of the old blocks, we analyzed their pattern of overlap. The degree of block overlap observed in the pseudo genome was compared with the degree of overlap that would be expected if all of the discovered blocks had been formed by random independent duplications. Simulation of random independent duplications was done by sequentially allocating each block pair to a random genomic location, except that overlaps between sister regions were not permitted. This procedure was repeated 10,000 times, and in each iteration, the total overlap between adjacent blocks was calculated and expressed as the number of genes shared between overlapping blocks. The proportion of the 10,000 random data sets with lower overlap than the pseudo genome is a direct estimate of the P value that can be attached to the hypothesis that all of the observed duplicated blocks came from random and independent segmental duplications. The degree of overlap between blocks in the pseudo genome (2533 genes) was significantly lower than in the simulations (mean overlap 3587 genes; SD 471; P = 0.0053), which indicates that at least some of the old blocks were formed by ancient polyploidy-type event(s).
Timing of the Duplication Events
We compared the levels of synonymous substitution seen in the recent and old Arabidopsis blocks to the levels seen in large sets of orthologous genes compared between Arabidopsis and various other plant species. Transcribed sequences corresponding to several species representing major plant taxa were downloaded from the TIGR gene index database (Quackenbush et al. 2000). We selected cotton (Gossypium hirsutum), which is classified in the Rosid II group like A. thaliana, barrel medic (Medicago truncatula; Rosid I), tomato (Lycopersicon esculentum; Asterid), and rice (Oryza sativa; a monocot). Complete transcript sequences from Brassica rapa, a crucifer closely related to A. thaliana, were also downloaded from GenBank. The phylogeny of this set of species is shown in Figure 4A (derived from Soltis et al. 1999). We identified putative orthogous relationships between these sequences and Arabidopsis genes by reciprocal best BLAST hit (see Methods) and estimated their levels of synonymous substitution. However, as it has been demonstrated that some rice genes have extremely high G+C content (Carels and Bernardi 2000; Wong et al. 2002a), which could drastically inflate Ks estimates, we only analyzed rice sequences with synonymous site G+C content below 60%.
Figure 4.

Position of the duplication events in the tree of life. (A) Phylogenetic relationships between the analyzed plant species. The arrow marks the probable position of the recent large-scale duplication event identified in this analysis. (B) Distribution of Ks values obtained from sets of orthologous sequences between Arabidopsis and selected plant species and between paralogous sequences in recent and old Arabidopsis blocks. The numbers of sequences orthologous to Arabidopsis are 252 for Brassica rapa, 1358 for cotton, 1741 for Medicago, 1465 for tomato, and 1026 for rice. The paralog Ks distributions are for 2617 and 576 gene pairs occurring in recent duplicated blocks, and blocks identified in the pseudo ancestral genome, respectively. The vertical lines depict the positions of the modes of the distributions obtained from Arabidopsis paralogous genes.
The Ks distributions for each species pair (Fig. 4B) show some skew toward high Ks values, which is probably partly attributable to wrongly defined ortholog pairs. We therefore compared the modes rather than the means of the distributions, because the mode is not affected by skew. For the comparisons of large numbers of orthologs between Arabidopsis and other species, the modal Ks values increase strictly with increasing phylogenetic distance, confirming the reliability of the approach. The Ks mode for Arabidopsis genes in recent sister regions (0.8–1.0 Ks) lies between the modes for the Arabidopsis-B. rapa comparison (0.4–0.6 Ks) and the Arabidopsis-cotton comparison (1.8–2.0 Ks).
Although the old blocks obviously result from multiple duplication events (Fig. 1), the distribution of their Ks values shows a single peak (mode, 2.0–2.2 Ks) similar in width to ortholog comparisons (Fig. 4), suggesting that a burst of gene duplications occurred in a short period of time. This observation is consistent with the possible large-scale duplication event suggested by the overlap analysis described above. The Ks mode for old blocks is similar to that for the Arabidopsis/Medicago ortholog comparison and slightly below that for Arabidopsis versus tomato. This suggests that a burst of gene duplications might have occurred at approximately the time of divergence between the Rosid I and Rosid II groups. It is notable that most of the Ks values obtained for duplicated genes are below the Arabidopsis/rice Ks distribution mode, indicating that most of the blocks identified in this work were formed after the monocot–dicot split.
DISCUSSION
We identified a group of 45 duplicated block pairs that are relatively young and homogeneous in age (Fig. 2). These blocks cover 70% of the genome without any significant overlap. Together, these two observations represent very strong evidence in favor of a recent polyploidy event followed by extensive chromosomal rearrangements. However, it should be pointed out that these blocks vary significantly from each other in terms of Ks distribution (Kruskal-Wallis test for among-block Ks variation; P < 0.001), suggesting that some pairs of recent sister regions might have slightly different ages of divergence. This could indicate that the Arabidopsis ancestor evolved through segmental allotetraploidy as already suggested for maize (Gaut and Doebley 1997).
Although more comprehensive, our map of recent blocks can be superimposed on maps proposed earlier (Arabidopsis Genome Initiative 2000; Blanc et al. 2000; Paterson et al. 2000; see http://www.tigr.org/tdb/e2k1/ath1/arabGenomeDups.html), confirming those studies. Most of the recently duplicated regions were also discovered by Vision et al. (2000). However, they estimated that these blocks fell into four different age classes ranging from 100–200 Mya, using a questionable dating strategy (Sankoff 2001; Wolfe 2001). In contrast, our analysis of Ks levels and the overlap pattern shows clearly that the recent event was a single polyploidy that encompassed the whole genome. Moreover, using levels of nonsynonymous substitutions in a small number of duplicated genes, the same group estimated that the more recent polyploidy event occurred ∼112 Mya (Ku et al. 2000). However, our comparison of between-species Ks distributions (Fig. 4) clearly places the date of the recent polyploidy event between the Arabidopsis–Brassica split and the Arabidopsis–cotton split, and definitively after the Arabidopsis–Medicago split, which is estimated at 92 Mya (Gandolfo et al. 1998; Grant et al. 2000). The median Ks value for Arabidopsis–Brassica rapa orthologs is 0.46, whereas that obtained from Arabidopsis recently duplicated blocks is 0.90. Thus, we can estimate roughly that the recent polyploidy event is twice as old as the Brassica–Arabidopsis split. The separation between Arabidopsis and Brassica lineages is estimated to be between 12 and 20 Mya (Yang et al. 1999; Acarkan et al. 2000; Koch et al. 2000, 2001), which places the polyploidy event around 24–40 Mya, probably close to the emergence of the crucifers. This estimate is substantially younger than the previous estimates by Vision et al. (2000). It is also lower than those of Lynch and Conery (2000) (65 Mya) and Simillion et al. (2002) (75 Mya), probably because those studies used more indirect calibrations of the rate of synonymous substitution. It is possible that differential losses of some genes duplicated before the separation between Arabidopsis and B. rapa could have led to wrongly defined orthologous pairs in our dataset (i.e., paralogs misidentified as orthologs). This would increase the median Ks value for the Arabidopsis–B. rapa comparison and ultimately result in underestimation of the ratio between the polyploidy date and the Arabidopsis–Brassica divergence date. Thus, our estimate of 24–40 Myr for the polyploidy must be regarded as a lower bound.
However, our estimate is in good agreement with the work of Galloway et al. (1998) who studied the phylogeny of 13 Brassicaceae species using arginine decarboxylase (Adc) nuclear genes. Most of the sampled crucifer species have orthologs of both of the two paralogous Arabidopsis Adc genes, which are located in a pair of recently duplicated blocks on chromosomes 2 and 4. Interestingly, the branching pattern of the resulting tree places the duplication of the Arabidopsis Adc genes, and by extrapolation, the polyploidy event, after the split between Arabidopsis and Aethionema grandiflora (one of the most basal crucifers), which is estimated at around 40 Mya (Koch et al. 2000, 2001). Thus, this suggests that the most recent polyploidy event is specific to the crucifer family, although not linked to their emergence.
It has been shown that the basal chromosome number of the Arabidopsis, Arabis, and Brassica genera is n = 8 (Koch et al. 1999), suggesting that their closest nonpolyploid ancestor was n = 4. Almost all analyzed species of Arabidopsis and Arabis sensu strictu closely related to A. thaliana have n = 8. The reduction to five chromosomes in the A. thaliana lineage dates to after the split between A. thaliana and A. halleri (Koch et al. 1999), a few million years ago. Thus, A thaliana obviously sustained the loss of three centromeres, possibly along with other limited chromosomal segment deletions and chromosome fusions, very recently. This may explain why only 80% of the Arabidopsis genome (excluding centromeric regions) is now seen to be duplicated. Moreover, whereas all centromeres can be paired in the paleopolyploid Saccharomyces cerevisiae genome (Wong et al. 2002b), the absence of detectable duplicated regions spanning Arabidopsis centromeres may partly be explained by three of the four potential pairs having been dissociated.
As well as the blocks derived from recent polyploidy, we also detected block pairs in Arabidopsis originating from older events, as evidenced by their higher Ks values (Fig. 2). By reconstructing the approximate gene order that existed before the recent polyploidy, we discovered a set of 68 pairs of old duplicated regions spanning 49% of the pseudo ancestral genome. Although these regions overlap extensively, indicating multiple duplication events, the degree of overlap is significantly lower than what would be expected if they had all been formed independently. Thus, it is very likely that some of the old sister regions were formed through an additional large-scale duplication event such as a polyploidy. Ks estimates suggest that most of these old blocks were formed between the dates of the Arabidopsis/cotton and monocot/dicot divergences (Fig. 4B). However, Ks is only statistically reliable for relatively recent events and shows an unexpectedly high variation among genes duplicated at the same time (Long et al. 2001; Zhang et al. 2002). The relatively high Ks values observed in old blocks make it impossible to estimate how many events occurred in this interval, or the relative contributions of polyploidy versus smaller duplications. Moreover, these circumstances hinder precise placement of old block duplications in the phylogeny using Ks (Fig. 4), although we can be confident that most of the old duplication events post-date the monocot–dicot divergence. Further analysis of these older event(s) will require alternative approaches such as phylogenetic analysis of protein sequences and will depend on the availability of orthologous plant sequences.
If we consider the evolution of the Arabidopsis genome since the more recent polyploidy event, it appears that independent duplications of chromosomal segments of >50 kb have been quite rare during the last ∼20 Myr; we did not detect any in our analysis. At the same time, many crucifer species from the Arabidopsis, Brassica, and Arabis genera are known polyploids (Koch et al. 1999), and even genetically diploid Brassica species genomes evolved through several rounds of polyploidy specific to this lineage (Lagercrantz and Lydiate 1996). To the extent that we can generalize from the recent history of crucifer genomes to more ancestral plants, we can speculate that all observed duplicated blocks in the A. thaliana genome originate from large-scale events. This would not be very surprising, as polyploidy is known to be very widespread in the plant kingdom, with at least 50%–70% of plant species being estimated to have experienced polyploidy in their ancestry (Wendel 2000) and 2%–4% of plant speciation events being attributed to polyploidy (Otto and Whitton 2000). The actual numbers might be even greater if we consider that old duplication events are relatively difficult to detect even with a complete genome sequence in hand, owing to rapid genomic changes, as exemplified by this study.
METHODS
Data Preparation
The annotated sequences of the five chromosomes of A. thaliana were downloaded from the Genomes Division of GenBank as five single files. The sequence accession/version numbers and GI numbers were NC_003070.1 (GI:15217430), NC_0003071.1 (GI:15224037), NC_003074.1 (GI:15228160), NC_003075.1 (GI:15233324), and NC_003076.1 (GI:15237134) for chromosomes 1 through 5, respectively. These sequence and annotation versions were dated August 13, 2001, and specify 25,542 predicted proteins. All-against-all protein sequence similarity searches were done using the ssearch34 program (Smith and Waterman 1981) with the seg filter (Wootton and Federhen 1996). Before searching for duplicated regions in the genome, we preprocessed the data to remove tandemly duplicated genes and transposable elements, because these could potentially lead to detection of spurious sister regions. Aligned proteins with an E-value ≤ 1e-20 and corresponding to sequences residing <15 genes apart on the chromosome were designated as tandem duplicated genes. For each tandem array, we retained only the longest sequence for further analysis. This filtering step reduced the proteome by 2960 proteins (11.6%), which is in accordance with the previous observation that 17% of Arabidopsis genes are organized in tandem arrays (Arabidopsis Genome Initiative 2000). We further filtered the database by discarding match information between sequences showing similarity to proteins annotated as transposable elements, which removed 788 proteins.
Block Detection
The approach used in this study has been already described by McLysaght et al. (2002) and was adapted for the case of Arabidopsis. In our analysis, a link was created between two similar genes if (1) the ssearch34 alignment between the corresponding proteins gave an E-value lower than 1e-20, (2) the E-value did not exceed 1e20 times the E-value of the best non-self-hit, in order to restrict the analysis to the closest family members, and (3) at least 50% of the longest sequence is aligned. (4) If the number of family members fulfilling these requirements was >20, the whole family was skipped. (5) Finally, a maximum of 30 unduplicated genes was allowed between 2 duplicated genes in each sister region.
Estimation of the Level of Synonymous Substitutions (Ks)
Protein sequences were aligned using the Smith-Waterman algorithm (Smith and Waterman 1981), and the resulting alignment was used as a guide to align the nucleotide sequences. After removing gaps, the level of synonymous substitutions was estimated using the maximum likelihood method implemented in codeml (Yang 1999) under the F3x4 model (Goldman and Yang 1994). We kept all Ks values up to 10, which allow reliable phylogenetic analysis (Yang 1998; Anisimova et al. 2001) (see also http://abacus.gene.ucl.ac.uk/software/pamlFAQs.html).
Ortholog Sequence Analysis
Plant transcribed nucleotide sequences from the TIGR gene index (Quackenbush et al. 2000) (http://www.tigr.org/tdb/tgi/) and GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) were searched against the Arabidopsis proteome using BLASTX (Altschul et al. 1997), and, conversely, the Arabidopsis proteins were searched successively against the transcribed sequence sets using TBLASTN. Two sequences were defined as orthologs when each of them was the best hit of the other. For Ks estimation, the plant transcribed sequences were translated using the Arabidopsis ortholog protein as a guide with the Genewise program (Birney et al. 1996).
WEB SITE REFERENCES
http://wolfe.gen.tcd.ie/athal/dup; Interactive maps of duplicated blocks (classified according to age classes) found using the BLASTP program (protein against proteins) in this present study.
http://www.tigr.org/tdb/e2k1/ath1/arabGenomeDups.html; Arabidopsis duplicated blocks found by the Arabidopsis Genome Initiative, 2000, using the Mummer (DNA against DNA) and TBLASTX (compares the conceptual 6-frame translations from two genomic DNA sequences) programs.
http://www.ncbi.nlm.nih.gov/Genbank/; GenBank.
http://www.tigr.org/tdb/tgi/; TIGR Gene Indices.
http://abacus.gene.ucl.ac.uk/software/pamlFAQs.html; CODEML program documentation.
Acknowledgments
This work was supported by Science Foundation Ireland and Enterprise Ireland.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E MAIL khwolfe@tcd.ie; FAX 353-1-679-8558.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.751803.
REFERENCES
- 1.Acarkan A., Rossberg, M., Koch, M., and Schmidt, R. 2000. Comparative genome analysis reveals extensive conservation of genome organization for Arabidopsis thaliana and Capsella rubella. Plant J. 23: 55-62. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anisimova M., Bielawski, J.P., and Yang, Z. 2001. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18: 1585-1592. [DOI] [PubMed] [Google Scholar]
- 4.Arabidopsis Genome Initiative 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796-815. [DOI] [PubMed] [Google Scholar]
- 5.Bevan M., Mayer, K., White, O., Eisen, J.A., Preuss, D., Bureau, T., Salzberg, S.L., and Mewes, H.W. 2001. Sequence and analysis of the Arabidopsis genome. Curr. Opin. Plant Biol. 4: 105-110. [DOI] [PubMed] [Google Scholar]
- 6.Birney E., Thompson, J.D., and Gibson, T.J. 1996. PairWise and SearchWise: Finding the optimal alignment in a simultaneous comparison of a protein profile against all DNA translation frames. Nucleic Acids Res. 24: 2730-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanc G., Barakat, A., Guyot, R., Cooke, R., and Delseny, M. 2000. Extensive duplication and reshuffling in the Arabidopsis genome. Plant Cell 12: 1093-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carels N. and Bernardi, G. 2000. Two classes of genes in plants. Genetics 154: 1819-1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galloway G.L., Malmberg, R.L., and Price, R.A. 1998. Phylogenetic utility of the nuclear gene arginine decarboxylase: An example from Brassicaceae. Mol. Biol. Evol. 15: 1312-1320. [DOI] [PubMed] [Google Scholar]
- 10.Gandolfo M.A., Nixon, K.C., and Crepet, W.L. 1998. New fossil flower from the Turonian of New Jersey: Dressiantha bicarpellata gen. et sp. nov. (Capparales). Am. J. Bot. 85: 964-974. [PubMed] [Google Scholar]
- 11.Gaut B.S. and Doebley, J.F. 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc. Natl. Acad. Sci. 94: 6809-6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldman N. and Yang, Z. 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11: 725-736. [DOI] [PubMed] [Google Scholar]
- 13.Grant D., Cregan, P., and Shoemaker, R.C. 2000. Genome organization in dicots: Genome duplication in Arabidopsis and synteny between soybean and Arabidopsis. Proc. Natl. Acad. Sci. 97: 4168-4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu X. and Huang, W. 2002. Testing the parsimony test of genome duplications: A counterexample. Genome Res. 12: 1-2. [DOI] [PubMed] [Google Scholar]
- 15.Gu X., Wang, Y., and Gu, J. 2002. Age distribution of human gene families shows significant roles of both large- and small-scale duplications in vertebrate evolution. Nat. Genet 31: 205-209. [DOI] [PubMed] [Google Scholar]
- 16.Kashkush K., Feldman, M., and Levy, A.A. 2002. Gene loss, silencing and activation in a newly synthesized wheat allotetraploid. Genetics 160: 1651-1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch M., Bishop, J., and Mitchell-Olds, T. 1999. Molecular systematics and evolution of Arabidopsis and Arabis. Plant Biology 1: 529-537. [Google Scholar]
- 18.Koch M., Haubold, B., and Mitchell-Olds, T. 2001. Molecular systematics of the Brassicaceae: Evidence from coding plastidic matK and nuclear Chs sequences. Am. J. Bot. 88: 534-544. [PubMed] [Google Scholar]
- 19.Koch M.A., Haubold, B., and Mitchell-Olds, T. 2000. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 17: 1483-1498. [DOI] [PubMed] [Google Scholar]
- 20.Ku H.M., Vision, T., Liu, J., and Tanksley, S.D. 2000. Comparing sequenced segments of the tomato and Arabidopsis genomes: Large-scale duplication followed by selective gene loss creates a network of synteny. Proc. Natl. Acad. Sci. 97: 9121-9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagercrantz U. and Lydiate, D.J. 1996. Comparative genome mapping in Brassica. Genetics 144: 1903-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lander E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- 23.Long M., Thornton, K., Zhang, L., Gaut, B.S., Vision, T.J., Lynch, M., and Conery, J.C. 2001. Gene duplication and evolution. Science 293: 1551a. [Google Scholar]
- 24.Lynch M. and Conery, J.S. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151-1155. [DOI] [PubMed] [Google Scholar]
- 25.McLysaght A., Hokamp, K., and Wolfe, K.H. 2002. Extensive genomic duplication during early chordate evolution. Nat. Genet. 31: 200-204. [DOI] [PubMed] [Google Scholar]
- 26.Otto S.P. and Whitton, J. 2000. Polyploid incidence and evolution. Annu. Rev. Genet. 34: 401-437. [DOI] [PubMed] [Google Scholar]
- 27.Paterson A.H., Bowers, J.E., Burow, M.D., Draye, X., Elsik, C.G., Jiang, C.X., Katsar, C.S., Lan, T.H., Lin, Y.R., Ming, R., et al. 2000. Comparative genomics of plant chromosomes. Plant Cell 12: 1523-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quackenbush J., Liang, F., Holt, I., Pertea, G., and Upton, J. 2000. The TIGR gene indices: Reconstruction and representation of expressed gene sequences. Nucleic Acids Res. 28: 141-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sankoff D. 2001. Gene and genome duplication. Curr. Opin. Genet. Dev. 11: 681-684. [DOI] [PubMed] [Google Scholar]
- 30.Simillion C., Vandepoele, K., Van Montagu, M.C.E., Zabeau, M., and Van de Peer, Y. 2002. The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad.Sci. 99: 13627-13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147: 195-197. [DOI] [PubMed] [Google Scholar]
- 32.Soltis P.S., Soltis, D.E., and Chase, M.W. 1999. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402: 402-404. [DOI] [PubMed] [Google Scholar]
- 33.Venter J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- 34.Vision T.J., Brown, D.G., and Tanksley, S.D. 2000. The origins of genomic duplications in Arabidopsis. Science 290: 2114-2117. [DOI] [PubMed] [Google Scholar]
- 35.Wendel J.F. 2000. Genome evolution in polyploids. Plant Mol. Biol. 42: 225-249. [PubMed] [Google Scholar]
- 36.Wolfe K.H. 2001. Yesterday's polyploids and the mystery of diploidization. Nat. Rev. Genet. 2: 333-341. [DOI] [PubMed] [Google Scholar]
- 37.Wolfe K.H. and Shields, D.C. 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708-713. [DOI] [PubMed] [Google Scholar]
- 38.Wong G.K., Wang, J., Tao, L., Tan, J., Zhang, J., Passey, D.A., and Yu, J. 2002a. Compositional gradients in Gramineae genes. Genome Res. 12: 851-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong S., Butler, G., and Wolfe, K.H. 2002b. Gene order evolution and paleopolyploidy in hemiascomycete yeasts. Proc. Natl. Acad. Sci. 99: 9272-9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wootton J.C. and Federhen, S. 1996. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 266: 554-571. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y.W., Lai, K.N., Tai, P.Y., and Li, W.H. 1999. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J. Mol. Evol. 48: 597-604. [DOI] [PubMed] [Google Scholar]
- 42.Yang Z. 1998. On the best evolutionary rate for phylogenetic analysis. Syst. Biol. 47: 125-133. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z., 1999. Phylogenetic analysis by maximum likelihood (PAML), version 2. University College, London, UK.
- 44.Zhang L., Vision, T.J., and Gaut, B.S. 2002. Patterns of nucleotide substitution among simultaneously duplicated gene pairs in Arabidopsis thaliana. Mol. Biol. Evol. 19: 1464-1473. [DOI] [PubMed] [Google Scholar]