

Albert et al. identify six measures which, in combination, predict progression to mild cognitive impairment with considerable accuracy. This should help identify individuals for inclusion in trials aimed at preclinical Alzheimer’s disease.
Keywords: preclinical Alzheimer’s disease, biomarkers, clinical trials
Abstract
Recent evidence indicates that measures from cerebrospinal fluid, MRI scans and cognitive testing obtained from cognitively normal individuals can be used to predict likelihood of progression to mild cognitive impairment several years later, for groups of individuals. However, it remains unclear whether these measures are useful for predicting likelihood of progression for an individual. The increasing focus on early intervention in clinical trials for Alzheimer’s disease emphasizes the importance of improving the ability to identify which cognitively normal individuals are more likely to progress over time, thus allowing researchers to efficiently screen participants, as well as determine the efficacy of any treatment intervention. The goal of this study was to determine which measures, obtained when individuals were cognitively normal, predict on an individual basis, the onset of clinical symptoms associated with a diagnosis of mild cognitive impairment due to Alzheimer’s disease. Cognitively normal participants (n = 224, mean baseline age = 57 years) were evaluated with a range of measures, including: cerebrospinal fluid amyloid-β and phosphorylated-tau, hippocampal and entorhinal cortex volume, cognitive tests scores and APOE genotype. They were then followed to determine which individuals developed mild cognitive impairment over time (mean follow-up = 11 years). The primary outcome was progression from normal cognition to the onset of clinical symptoms of mild cognitive impairment due to Alzheimer’s disease at 5 years post-baseline. Time-dependent receiver operating characteristic analyses examined the sensitivity and specificity of individual measures, and combinations of measures, as predictors of the outcome. Six measures, in combination, were the most parsimonious predictors of transition to mild cognitive impairment 5 years after baseline (area under the curve = 0.85; sensitivity = 0.80, specificity = 0.75). The addition of variables from each domain significantly improved the accuracy of prediction. The incremental accuracy of prediction achieved by adding individual measures or sets of measures successively to one another was also examined, as might be done when enrolling individuals in a clinical trial. The results indicate that biomarkers obtained when individuals are cognitively normal can be used to predict which individuals are likely to develop clinical symptoms at 5 years post-baseline. As a number of the measures included in the study could also be used as subject selection criteria in a clinical trial, the findings also provide information about measures that would be useful for screening in a clinical trial aimed at individuals with preclinical Alzheimer’s disease.
Introduction
Accumulating evidence indicates that the underlying neuropathological mechanisms associated with Alzheimer’s disease begin a decade or more before the emergence of cognitive impairment (Sperling et al., 2011). This understanding has had a substantial impact on the conduct of clinical trials related to Alzheimer’s disease, since it is hypothesized that disease-modifying therapies are likely to be more successful when administered early in the course of disease. Several clinical trials are currently underway among asymptomatic individuals known to be in the preclinical phase of Alzheimer’s disease, due to the presence of genetic mutations that cause Alzheimer’s disease (Moulder et al., 2013; Fleisher et al., 2015). A small number of trials have also been initiated among cognitively normal individuals thought to be at risk for progression to mild cognitive impairment (MCI), by virtue of their apolipoprotein E (APOE) genetic status (Reiman et al., 2011) or brain imaging evidence of amyloid accumulation (Sperling et al., 2014), one of the pathological hallmarks of Alzheimer’s disease (Holtzman, 2011). Moreover, many clinical trials are ongoing, or recently completed, that include individuals in the MCI phase of disease (Lasser et al., 2015; Sevigny et al., 2015). The recent failure of several therapeutic agents emphasizes the importance of not only finding improved medications for Alzheimer’s disease, but also of designing subject selection criteria that maximize the enrolment of subjects who are most likely to progress over the duration of the study, since lack of progression limits the ability to determine if a treatment is efficacious.
A number of prior studies have indicated that CSF and MRI Alzheimer’s disease biomarkers are associated with the risk of progression from normal cognition to MCI or dementia. These biomarkers include CSF amyloid-β, total tau and phosphorylated tau (p-tau) (Moghekar et al., 2013; Roe et al., 2013; Toledo et al., 2014; Vos et al., 2016), as well as the volumes of the hippocampus or entorhinal cortex on MRI (Csernansky et al., 2005; Toledo et al., 2014; Soldan et al., 2015). However, few studies are available that provide data on optimizing subject selection criteria for the preclinical phase of Alzheimer’s disease. This is because most longitudinal studies that have enrolled cognitively normal individuals and collected relevant measures have limited follow-up. Importantly, studies with limited follow-up tend to lack a sufficient number of clinical outcomes (i.e. number of cases who progress to MCI) necessary for robust statistical analyses to determine which measures most reliably predict progression.
Such analyses are feasible using data from the BIOCARD study, in which participants were cognitively normal when first enrolled, a wide range of informative measures were collected at baseline, and participants have now been followed for up to 20 years. The measures collected include: CSF, MRI, cognitive testing, and APOE genetic status. The availability of these measures at baseline, when the subjects were cognitively normal, and the unusually long duration of follow-up in the study (mean = 11 years), allowed the examination of several questions of particular relevance to the outcome of individuals with preclinical Alzheimer’s disease, and the design of clinical trials aimed at this phase of disease.
The primary goal of these analyses was to identify which measures, or combination of measures, obtained among individuals who were cognitively normal at enrolment, could be used to accurately predict, on an individual basis, subsequent progression from normal cognition to onset of clinical symptoms associated with a diagnosis of MCI due to Alzheimer’s disease. As a number of the measures included in the study could also be used as subject selection criteria in a clinical trial, we also examined the incremental accuracy of the prediction that could be achieved when adding individual measures, or sets of measures, successively to one another, as might be done when enrolling individuals in a clinical trial. Time-dependent receiver operating characteristic (ROC) analyses were used to evaluate the diagnostic accuracy, sensitivity, and specificity of the measures in predicting which individual subjects developed clinical symptoms associated with MCI due Alzheimer’s disease at different durations of follow-up (i.e. at 5, 7 and 10 years post-baseline).
Materials and methods
Study design
The BIOCARD study, the parent study from which these data are drawn, was initiated at the National Institutes of Health (NIH) in 1995. While at the NIH, subjects were administered a neuropsychological battery and clinical assessments annually. MRI scans, CSF, and blood specimens were obtained approximately every 2 years. The study was stopped in 2005 for administrative reasons and re-established at Johns Hopkins in 2009, at which point the annual clinical and neuropsychological assessments were reinitiated. Bi-annual collection of CSF and MRI scans was re-established in 2015, as well as the acquisition of PET scans using Pittsburgh Compound B (PiB) (see Supplementary Fig. 1 for a schematic representation of the study design).
Selection of participants
Recruitment was conducted by the staff of the Geriatric Psychiatry branch of the intramural program of the National Institute of Mental Health. At baseline, all participants completed a comprehensive evaluation at the NIH, consisting of a physical and neurological examination, an ECG, standard laboratory studies, and neuropsychological testing. Individuals were excluded from participation if they were cognitively impaired, or had significant medical problems such as severe cerebrovascular disease, epilepsy or alcohol or drug abuse.
A total of 349 individuals were initially enrolled in the study, after providing written informed consent. By design, ∼75% of the participants had a first degree relative with dementia of the Alzheimer type. The analyses presented here are based on data from 224 subjects who were cognitively normal at baseline and had complete observations on the baseline variables of interest. Most of the exclusions pertained to the availability of a complete dataset for the participants (see Supplementary material, section 1 for the reasons subjects were excluded from the analyses).
Of the 224 subjects included in these analyses, 178 subjects remained cognitively normal at their last visit (this includes 22 subjects with a diagnosis of ‘Impaired Not MCI’ at their last visit) and 46 subjects were diagnosed with MCI or dementia due to Alzheimer’s disease by the time of their last visit. The demographic characteristics of the subjects in the analysis are shown in Table 1, which are similar to the characteristics of the cohort as a whole.
Table 1.
Baseline characteristics of the participants included in the analyses in comparison to the cohort as a whole
| Variable | Cohort as a whole (n = 349) | Subjects in analyses (n = 224) |
|---|---|---|
| Age, mean years (SD) | 57.3 (10.4) | 56.9 (8.4) |
| Gender, % females | 57.6% | 62.1% |
| Education, mean years (SD) | 17.0 (2.4) | 17.1 (2.3) |
| Ethnicity, % Caucasians | 97.1 | 97.8% |
| % APOE4 carriers | 33.6 | 37.5 |
| MMSE, mean score (SD) | 29.5 (0.9) | 29.4 (1.0) |
| NART, mean score (SD) | 119.6 (7.9) | 121.0 (7.3) |
MMSE = Mini-Mental State Examination; NART = National Adult Reading Test.
Consensus diagnostic procedures
Clinical and cognitive assessments were completed annually at the NIH initially and subsequently at Johns Hopkins, as noted above. A consensus diagnosis for each study visit was established by the staff of the BIOCARD Clinical Core at Johns Hopkins (prospectively for subjects evaluated starting in 2009 and retrospectively for subjects evaluated at the NIH). As previously described (Albert et al., 2014), each consensus diagnosis was handled in a similar manner. First a syndromic diagnosis was established: (i) clinical data pertaining to the medical, neurologic and psychiatric status of the subject were examined; (ii) reports of changes in cognition by the subject and by a collateral source were reviewed; and (iii) decline in cognitive performance, based on review of longitudinal testing from multiple domains (and by comparison with published norms) was determined. If a subject was deemed to be impaired, the decision about the likely aetiology of the syndrome was based on the medical, neurological, and psychiatric information collected at each visit, as well as medical records obtained from the subject, where necessary. More than one aetiology could be endorsed for each subject (e.g. Alzheimer’s disease and vascular disease). The consensus diagnosis procedures followed the diagnostic recommendations incorporated in the NIA-AA working group reports for the diagnosis of MCI (Albert et al., 2011) and dementia due to Alzheimer’s disease (McKhann et al., 2011).
The estimated age of onset of clinical symptoms was established separately, based primarily on a semi-structured interview with the subject and the collateral source. The staff conducting the consensus diagnoses were blinded to the CSF and MRI measures and to the APOE status of the participants (see Supplementary material, section 2 for additional details regarding the diagnostic procedures).
Selection criteria for variables included in the analyses
The ROC analyses presented here include variables from the four primary domains evaluated in the BIOCARD study, obtained when subjects were first enrolled. These domains included: (i) CSF values; (ii) MRI measures; (iii) cognitive test scores; and (iv) APOE genetic status. To be as parsimonious as possible, we based the selection of which specific variables should be included in the ROC analyses on findings from prior publications in which we had conducted Cox regression analyses designed in a parallel fashion. In each of these prior publications we used Cox regression procedures to examine the relationship between the values obtained at baseline (when participants were cognitively normal) and time to onset of clinical symptoms consistent with a diagnosis of MCI due to Alzheimer’s disease. Since the measures in these prior analyses had been standardized (using z-scores), it was possible to not only determine the relationship between the baseline measure and the outcome of interest (onset of clinical symptoms) but to also directly compare the hazard ratios across variables and domains. The measures selected from these prior analyses are described below.
Cognitive assessments
The annual, comprehensive neuropsychological battery covered all major cognitive domains, including memory, executive function, language, visuospatial ability, attention, speed of processing and psychomotor speed (see Albert et al., 2014 for the complete battery). Of the 17 variables selected from the cognitive battery (based on exploratory plots of change patterns over time), nine were significantly associated with the outcome, i.e. time to onset of clinical symptoms (Albert et al., 2014). The majority of the significant associations pertained to tests of episodic memory. We selected the two cognitive measures with the strongest association between baseline and outcome, based on hazard ratios, to include in the ROC analyses: (i) Digit Symbol Substitution Test from the Wechsler Adult Intelligence Scale – Revised; and (ii) Verbal Paired Associates – Immediate recall from the Wechsler Memory Scale – Revised.
CSF assessments
The CSF specimens collected from the participants were analysed using the xMAP-based AlzBio3 kit (Innogenetics) run on the Bioplex 200 system. The assay procedures were identical to those used in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). CSF specimens were analysed in triplicate on the same plate. Three variables were generated from these analyses: (i) CSF amyloid-β; (ii) CSF total tau; and (iii) CSF p-tau (see Supplementary material, section 3 for further details of the CSF assays). We selected the two CSF measures that showed a significant association between baseline and time to onset of clinical symptoms, based on hazard ratios, to include in the ROC analyses: (i) CSF amyloid-β; and (ii) CSF p-tau (Moghekar et al., 2013). Although the ratios for CSF tau/amyloid-β and CSF p-tau/amyloid-β were also significantly associated with time to onset of clinical symptoms in our prior analyses (Moghekar et al., 2013), the use of the individual CSF measures allowed us to examine their incremental predictive value on an individual basis.
MRI assessments
The MRI scans acquired from the participants were obtained using a standard multi-modal protocol with a GE 1.5 T scanner. We used the coronal scans to reconstruct the volumes of the entorhinal cortex, the hippocampus and the amygdala, as well as the thickness of the entorhinal cortex. The coronal scans used an SPGR (spoiled gradient echo) sequence (repetition time = 24 ms, echo time = 2 ms, field of view = 256 × 256, thickness/gaP = 2.0/0.0 mm, flip angle = 20°, 124 slices). The scans were processed with a semi-automated method, using region of interest large deformation diffeomorphic metric mapping (ROI-LDDMM) techniques (Miller et al., 2013) (see Supplementary material, section 4 for further details of these methods). We selected the two MRI measures that showed a significant association between baseline and time to onset of clinical symptoms, based on hazard ratios, to include in the ROC analyses: (i) right entorhinal cortex thickness; and (ii) right hippocampal volume (normalized by intracranial cavity volume) (Soldan et al., 2015).
APOE genotype
APOE genotypes were determined by restriction endonuclease digestion of polymerase chain reaction amplified genomic DNA (performed by Athena Diagnostics). APOE4 carrier status was coded by an indicator variable, with E4 carriers coded as 1 if the subject had at least one E4 allele and non-carriers coded as 0.
Summary of variables included in the analyses
The variables included in the ROC analyses, based on the selection criteria described above, were therefore as follows: (i) the Digit Symbol Substitution test and Paired Associates Immediate Recall scores from the cognitive domain; (ii) CSF amyloid-β and CSF p-tau from the CSF domain; (iii) right hippocampal volume and right entorhinal cortex thickness from the MRI domain; and (iv) APOE4 status from the genetics domain. Aside from the variables described above, all of the ROC analyses always included demographic variables (age, education), since all prior analyses indicated that these variables have important modifying effects on time to onset of clinical symptoms.
Each of the variables described above were continuous variables (with the exception of APOE status, which was a binary measure). All continuous measures were standardized (using z-scores) prior to inclusion in the ROC analyses. This makes it possible to directly compare the hazard ratios from each variable to one another (note that the hazard ratio for APOE is therefore not comparable to the hazard ratios for the continuous variables).
Table 2 presents the means and standard deviations of the measures in the analysis for subjects who remained normal over time versus those who progressed to MCI; the P-values are based on t-tests or chi squares comparing the subjects in the two groups.
Table 2.
Mean and standard deviation of variables at baseline for subjects who remained normal versus subjects who developed clinical symptoms and were diagnosed with MCI or Alzheimer’s disease dementia on follow-up
| Variable | Remained normal (n = 178) | Progressed to MCI or Alzheimer’s disease dementia (n = 46) | P-values |
|---|---|---|---|
| Age | 56.5 (7.2) | 62.3 (11.4) | 0.002* |
| Gender, % female | 62.4 | 60.9 | 0.988 |
| Education | 17.1 (2.6) | 16.6 (2.4) | 0.193 |
| Ethnicity, % Caucasian | 99.4 | 91.3 | <0.01* |
| % APOE4 carriers | 36.0 | 43.5 | 0.442 |
| Digit Symbol Substitution | 55.5 (11.1) | 46.6 (8.1) | <0.001* |
| Paired Associates Immediate | 20.9 (2.8) | 19.1 (3.2) | <0.001* |
| CSF amyloid-β | 415.5 (93.8) | 363.8 (102.9) | 0.003* |
| CSF p-tau | 34.1 (12.7) | 44.6 (21.8) | 0.003* |
| R. Hippocampus volume (mm3) | 1.70 (0.21) | 1.66 (0.23) | 0.318 |
| R. entorhinal cortex thickness (mm) | 2.14 (0.30) | 2.00 (0.26) | 0.002* |
R = right.
*Significant difference between groups (P < 0.05).
Statistical analysis
The overall goal of the time-dependent ROC analyses, as noted above, was to evaluate the prognostic accuracy of the measures described above in predicting which individual subjects developed clinical symptoms consistent with a diagnosis of MCI due to Alzheimer’s disease. These analyses were conducted with three durations of follow-up: 5, 7 and 10 years post-baseline. Four sets of ROC analyses were performed, using these time frames. First, we examined the predictability of the measures for each individual domain. Second, we examined the predictability of all the variables combined (i.e. the Full Model). Third, we sought to determine if a smaller set of measures would have comparable results to the Full Model, reducing the number of variables that would need to be assessed (i.e. referred to here as the Efficient Model); to accomplish this goal, variables were selected on the basis that each one had to be statistically significant when combined together, based on hazard ratios. Lastly, we examined the incremental predictability of variables in models designed to emulate selection criteria that might be used in clinical trials aimed at individuals with preclinical Alzheimer’s disease.
Demographic variables (age, education) were included in all models. For each model mentioned above, we first combined the relevant variables by entering them into a Cox proportional hazards model. The next step was to assess model fit. Cox models were run for every potential combination of variables (among the set of variables chosen for that particular model) in order to determine whether a model with all variables—or a model with a reduced set of variables—produced a better model fit. The Akaike Information Criterion (AIC) was used to compare models to one another to determine the model fit for a given set of biomarker variables and covariates. The AIC was selected for this purpose because it provides an index of the relative balance of model fit (based on the partial likelihood function for the Cox proportional hazards model) and model parsimony (based on the number of parameters in the model). A smaller AIC value indicates a better balance between fit and parsimony (Akaike, 1974).
If the AIC criterion for a set of measures was acceptable (i.e. the difference between the alternate models was <2), then the partial likelihood method for the Cox proportional hazards model was used to create a weighted sum of the measures (the weights being the log hazard ratio corresponding to each measure). Next, the weighted combination of measures from the proportional hazards model was used so that the area under the ROC curve (AUC) was maximized (McIntosh and Pepe, 2002; Blanche et al., 2013). The ROC represents a combined function of the sensitivity (true positive rate) and the specificity (true negative rate) of prediction and the AUC is widely considered a highly informative reflection of a measure(s) overall accuracy for predicting a disease-related outcome. The optimal sensitivity and specificity cut-off point for each model was established by maximizing the Youden index (sensitivity + specificity − 1) (Youden, 1950). The combined set of markers with the higher AUC was considered to be more predictive of disease progression. In this setting the AUC measures the intrinsic ability of the variables to discriminate between participants who developed clinical symptoms and participants who remained normal.
Lastly, different models were compared to one another using point-wise confidence intervals of the AUCs, with confidence intervals constructed using the bootstrap method (Hilbe, 2011). All analyses were implemented in R, Version 3.1.0 (see Supplementary material, section 5 for further details regarding the statistical methods).
Results
Predicting progression from normal cognition to MCI for individual domains
The first set of ROC analyses assessed the predictability of the measures from each individual domain separately (i.e. CSF, MRI, Cognition, APOE status). For each domain, the optimal AIC value (i.e. lowest) was obtained when including both variables from the given domain in the model (after co-varying age and education). As shown in Table 3, in the optimal model for each domain, each of the individual variables were significantly associated with progression from normal cognition to the onset of symptoms of MCI, except for CSF amyloid-β, which was not significant (P = 0.057). Table 3 also shows the AUCs, sensitivities and specificities for the predictability of each domain in relation to the outcome. AUCs were ∼0.70 for all domains, indicating moderate predictive power.
Table 3.
Predicting progression from normal cognition to MCI using measures from individual domains
| Variable | HR of model (95% CI) | HR: P-value | AUC of Model (95% CI) | Time to outcome for model | Model sensitivity | Model specificity |
|---|---|---|---|---|---|---|
| APOE4 | 0.703 (0.627, 0.798) | 5 years | 0.629 | 0.660 | ||
| 0.699 (0.628, 0.784) | 7 years | 0.613 | 0.671 | |||
| 0.685 (0.617, 0.764) | 10 years | 0.618 | 0.644 | |||
| APOE4a | 1.862 (1.013, 3.424) | 0.045 | ||||
| Cognitive domain | 0.764 (0.715, 0.834) | 5 years | 0.690 | 0.705 | ||
| 0.768 (0.720, 0.831) | 7 years | 0.684 | 0.718 | |||
| 0.767 (0.712, 0.831) | 10 years | 0.675 | 0.724 | |||
| Paired Associates Immediate | 0.630 (0.485, 0.819) | 0.001 | ||||
| Digit Symbol Substitution | 0.550 (0.381, 0.795) | 0.001 | ||||
| MRI domain | 0.740 (0.679, 0.819) | 5 years | 0.641 | 0.710 | ||
| 0.722 (0.670, 0.788) | 7 years | 0.662 | 0.659 | |||
| 0.705 (0.652, 0.773) | 10 years | 0.616 | 0.678 | |||
| R. Hippocampus volume | 0.728 (0.552, 0.961) | 0.025 | ||||
| R. entorhinal cortex thickness | 0.668 (0.492, 0.905) | 0.009 | ||||
| CSF domain | 0.717 (0.664, 0.812) | 5 years | 0.572 | 0.750 | ||
| 0.714 (0.663, 0.788) | 7 years | 0.578 | 0.735 | |||
| 0.740 (0.681, 0.807) | 10 years | 0.549 | 0.816 | |||
| Amyloid-β | 0.765 (0.581, 1.008) | 0.057 | ||||
| P-tau | 1.391 (1.069, 1.812) | 0.014 | ||||
aAPOE4 is a binary variable, and thus not standardized as other continuous variables. Therefore its hazard ratio is not comparable to those of continuous variables.
HR = hazard ratio; R = right. Age and education were entered first in each model.
Predicting progression from normal cognition to MCI combining variables from multiple domains
The second set of ROC analyses assessed the accuracy of predicting progression from normal cognition to MCI using the Full Model, which combined all of the variables in the analysis, with no prespecified ordering of the variables. This model was associated with high predictive accuracy (AUC > 0.83 at 5, 7, and 10 years post-baseline) (see Table 4 for the hazard ratios, AUCs, sensitivities and specificities).
Table 4.
Predicting progression from normal cognition to MCI using variables from multiple domains: the Full Model, Efficient Model, and Demographics only Model
| Variable | HR of model (95% CI) | HR: P-value | AUC of model (95% CI) | Time to outcome for model | Model sensitivity | Model specificity |
|---|---|---|---|---|---|---|
| Full Model | 0.850 (0.807, 0.913) | 5 years | 0.804 | 0.740 | ||
| 0.843 (0.803, 0.897) | 7 years | 0.815 | 0.724 | |||
| 0.831 (0.781, 0.890) | 10 years | 0.764 | 0.759 | |||
| Age | 1.364 (1.014, 1.834) | 0.040 | ||||
| Education | 0.750 (0.535, 1.052) | 0.095 | ||||
| APOE4a | 1.904 (1.024, 3.541) | 0.042 | ||||
| Paired Associates Immediate | 0.617 (0.469, 0.812) | 0.001 | ||||
| Digit Symbol Substitution | 0.454 (0.315, 0.655) | <0.001 | ||||
| CSF amyloid-β | 0.785 (0.572, 1.077) | 0.133 | ||||
| CSF p-tau | 1.779 (1.355, 2.336) | <0.001 | ||||
| R. Hippocampus volume | 0.699 (0.526, 0.930) | 0.014 | ||||
| R. entorhinal cortex Thickness | 0.594 (0.429, 0.821) | 0.002 | ||||
| Efficient Model | 0.849 (0.802, 0.910) | 5 years | 0.799 | 0.745 | ||
| 0.843 (0.798, 0.897) | 7 years | 0.803 | 0.735 | |||
| 0.822 (0.769, 0.886) | 10 years | 0.737 | 0.770 | |||
| Age | 1.430 (1.069, 1.914) | 0.016 | ||||
| Education | 0.737 (0.519, 1.045) | 0.086 | ||||
| APOE4a | 2.068 (1.124, 3.805) | 0.020 | ||||
| Paired Associates Immediate | 0.625 (0.476, 0.821) | 0.001 | ||||
| Digit Symbol Substitution | 0.445 (0.306, 0.648) | <0.001 | ||||
| CSF amyloid-β | - | - | ||||
| CSF p-tau | 1.912 (1.490, 2.454) | <0.001 | ||||
| R. Hippocampus volume | 0.667 (0.503, 0.884) | 0.005 | ||||
| R. entorhinal cortex thickness | 0.588 (0.424, 0.815) | 0.001 | ||||
| Demographics Model | 0.681 (0.614, 0.770) | 5 years | 0.592 | 0.675 | ||
| 0.678 (0.615, 0.753) | 7 years | 0.585 | 0.676 | |||
| 0.680 (0.612, 0.756) | 10 years | 0.566 | 0.701 | |||
| Age | 1.079 (1.045, 1.113) | <0.01 | ||||
| Education | 0.909 (0.804, 1.029) | 0.132 | ||||
aAPOE4 is a binary variable, and thus not standardized as other continuous variables. Therefore its hazard ratio is not comparable to those of continuous variables.
HR = hazard ratio; R = right.
We then examined the hazard ratio for each variable in the Full Model to determine if it was statistically significant. The only variable that did not meet these criteria was CSF amyloid-β, as noted above. This variable was therefore excluded, and the Efficient Model was rerun with the remaining variables. The predictive accuracy of the Efficient Model was also high (AUC > 0.82 at 5, 7, and 10 years post-baseline) (Table 4). Statistical comparisons between the Full Model and the Efficient Model revealed no significant differences. Specifically, the high AUCs for both the Full and Efficient Models (>0.83 and >0.82, respectively) did not differ significantly, indicating no difference in predictability (P = 0.53, 0.63, and 0.16, at 5, 7 and 10 years, respectively). This finding is illustrated graphically in Fig. 1, which shows that the Full Model and the Efficient Model have overlapping time-dependent ROC curves at 5 years post-baseline. Likewise, the difference in AIC values between the Full and Efficient Models (393.99 and 394.25, respectively) was <2 at 5 years post-baseline, indicating that the two models were indistinguishable from one another at this time frame (Hilbe, 2011).
Figure 1.
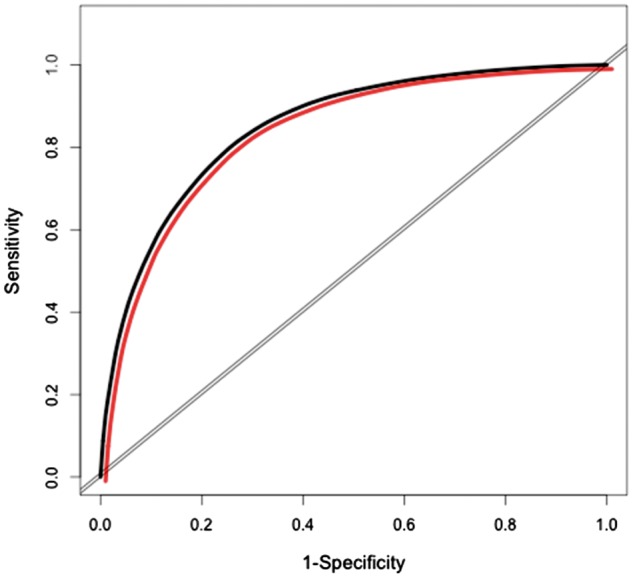
ROC curves for Full Model and Efficient Model. Time dependent ROC curves for the Full Model and the Efficient Model at 5 years post-baseline. Black = Full Model; red = Efficient Model.
Of note, an examination of cases that were misclassified by the Efficient Model at 5 years post-baseline revealed that about 35% of false positive classifications pertained to individuals who progressed at a later time point (mean time from baseline to symptom onset = 7.9 years) (Supplementary material, section 6).
Since the analyses of both the Full Model and the Efficient model demonstrated the importance of demographic variables for accurate prediction (Table 4), we conducted separate ROC analyses with only these variables. The AUCs for demographics alone were approximately 0.68 for all time frames post-baseline (Table 4).
Increment in predictability for variables added in order of potential application in a clinical trial
The last set of ROC analyses examined models designed to emulate an approach to screening that might be used in a clinical trial. In the first model, APOE status was included first, immediately after the demographic variables – this model was designed to emulate the situation in which APOE4 carrier status might be used as an inclusion criterion in a clinical trial aimed at individuals with preclinical Alzheimer’s disease (such as in the API Trial) (Reiman et al., 2011). We then examined the increment in predictability as measures were added consecutively within the model, based on feasibility in a clinical trial, to determine how much each domain adds to the accuracy of prediction at each time point (i.e. APOE status, cognitive measures, MRI measures and CSF measures, adjusted by demographics). Similar to the analyses described above, we continued to require that all variables added to the model had to have a significant hazard ratio and that the model had the smallest AIC compared to alternative models.
Table 5 shows the hazard ratio for each variable when added to the model (as well as the AUCs, sensitivities and specificities) for predicting onset of clinical symptoms at 5, 7 and 10 years post-baseline. Table 5 also shows the P-values comparing the AUCs for models in which we incrementally added variables from the cognitive, MRI and CSF domains, respectively. With only APOE status and demographics in the model, the AUC was approximately 0.70 at 5, 7 and 10 years post-baseline. The addition of the cognitive variables to this model significantly increased the AUC to approximately 0.78 for all follow-up time points (all P < 0.03). The addition of the MRI measures marginally improved predictability above and beyond the genetic and cognitive domains at 5 years (P = 0.052), but the addition of the MRI measures did not significantly improve predictability at 7 and 10 years post-baseline. However, predictability was significantly increased by the addition of CSF p-tau at the last step, increasing AUCs to approximately 0.84 (P < 0.03 for each time point). Figure 2 shows the incremental change in the time-dependent ROC curves predicting the onset of clinical symptoms for variables in this model at 5 years post-baseline. Of note, in a separate set of analyses, using the same model, we added the ratio of CSF p-tau/amyloid-β at the last step (instead of CSF p-tau alone) and found that the addition of this measure did not significantly increase predictability. Moreover, the same finding was true if we added the ratio of CSF.p-tau/amyloid-β at the last step, but APOE4 status was not included at the first step (Supplementary material, section 6).
Table 5.
Increment in prediction of progression from normal cognition to MCI for variables added in order of potential application in a clinical trial
| Variable | HR of model (95% CI) | HR: P-value | AUC of model (95% CI) | Time to outcome for model | Model sensitivity | Model specificity | Change in AUC versus prior step in model: P-value |
|---|---|---|---|---|---|---|---|
| APOE4 | 0.703 (0.627, 0.798) | 5 years | 0.629 | 0.660 | - | ||
| 0.699 (0.628, 0.784) | 7 years | 0.613 | 0.671 | - | |||
| 0.685 (0.617, 0.764) | 10 years | 0.618 | 0.644 | - | |||
| APOE4a | 1.862 (1.013, 3.424) | 0.045 | |||||
| APOE4+ Cognitive | 0.777 (0.730, 0.850) | 5 years | 0.708 | 0.710 | 0.021 | ||
| 0.785 (0.738, 0.852) | 7 years | 0.696 | 0.735 | 0.006 | |||
| 0.772 (0.716, 0.842) | 10 years | 0.680 | 0.724 | 0.010 | |||
| APOE4a | 1.931 (1.042, 3.580) | 0.037 | |||||
| Paired Associates Immediate | 0.629 (0.484, 0.816) | <0.001 | |||||
| Digit Symbol Substitution | 0.553 (0.384, 0.796) | 0.001 | |||||
| APOE4 + Cognitive + MRI | 0.811 (0.769, 0.880) | 5 years | 0.723 | 0.750 | 0.052 | ||
| 0.811 (0.769, 0.871) | 7 years | 0.723 | 0.747 | 0.062 | |||
| 0.787 (0.733, 0.861) | 10 years | 0.680 | 0.759 | 0.223 | |||
| APOE4a | 2.036 (1.108, 3.740) | 0.022 | |||||
| Paired Associates Immediate | 0.695 (0.534, 0.905) | 0.007 | |||||
| Digit Symbol Substitution | 0.500 (0.347, 0.719) | <0.001 | |||||
| R. Hippocampus volume | 0.712 (0.539, 0.939) | 0.016 | |||||
| R. entorhinal cortex thickness | 0.705 (0.522, 0.952) | 0.022 | |||||
| APOE4 + Cognitive + MRI + CSF p-tau | 0.849 (0.802, 0.910) | 5 years | 0.799 | 0.745 | 0.015 | ||
| 0.843 (0.798, 0.897) | 7 years | 0.803 | 0.735 | 0.014 | |||
| 0.822 (0.769, 0.886) | 10 years | 0.737 | 0.770 | 0.024 | |||
| APOE4a | 2.068 (1.124, 3.805) | 0.020 | |||||
| Paired Associates Immediate | 0.625 (0.476, 0.821) | 0.001 | |||||
| Digit Symbol Substitution | 0.445 (0.306, 0.648) | <0.001 | |||||
| R. Hippocampus volume | 0.667 (0.503, 0.884) | 0.005 | |||||
| R. entorhinal cortex thickness | 0.588 (0.424, 0.815) | 0.001 | |||||
| CSF p-tau | 1.912 (1.490, 2.454) | <0.001 | |||||
aAPOE4 is a binary variable, and thus not standardized as other continuous variables. Therefore its hazard ratio (HR) is not comparable to those of continuous variables.
R = right.
Age and education were entered first in each model.
Figure 2.
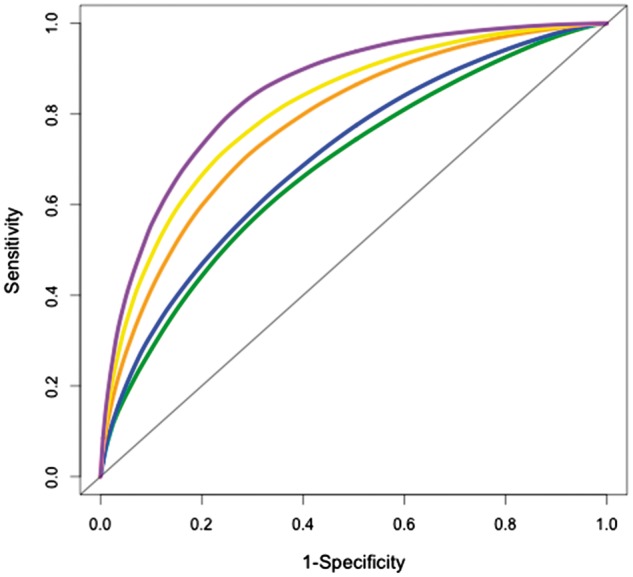
ROC Curves ordered by applicability in a clinical trial. Time-dependent ROC curves for measures in the Efficient Model ordered by applicability in a clinical trial at 5 years post-baseline. Purple: demographics, APOE4, Cognitive, MRI, CSF; yellow: demographics, APOE4, Cognitive, MRI; orange: demographics, APOE4, Cognitive; blue: demographics, APOE4; green: demographics.
In the second and third models within this set we focused primarily on the predictability derived from the introduction of the first variable(s) in the model (after the demographics). Since amyloid imaging using PET is currently being used to screen subjects for inclusion in a clinical trial of cognitively normal individuals (e.g. the A4 Study) (Sperling et al., 2014), the second model examined the impact of putting CSF amyloid-β in the model first. With only CSF amyloid-β and demographics in the model, the AUC ranged from 0.70 to 0.72 at 5, 7 and 10 years post-baseline. The sensitivity was 0.64 and the specificity was 0.67 at 5 years. With the addition of other domains to the model, the results were comparable to those in which APOE was added first (Supplementary material, section 7).
In the third model, CSF amyloid-β and CSF p-tau were included first, after the demographic variables—this model was designed to anticipate a future study in which both amyloid imaging and tau imaging might be used to screen subjects for inclusion in a clinical trial aimed at randomizing those who were both amyloid and tau PET positive. With CSF amyloid-β and CSF p-tau in the model (after demographics), the AUC ranged from 0.72 to 0.74 at 5, 7 and 10 years post-baseline (see Table 3, showing model results for the CSF domain). The sensitivity was 0.57 and the specificity was 0.75 at 5 years post-baseline. With the addition of the cognitive and MRI domains to the model, there was an increase in the AUC, with results comparable to those in which either APOE or CSF amyloid-β was added first (data not shown).
As noted above, the models in which two cognitive tests or two MRI variables were included, after the demographic variables, also showed moderate predictability (see Table 3, showing model results for the cognitive and MRI domains). Screening procedures such as these are often considered when more technologically complex and costly methods, such as PET scanning, are not feasible. Of note, though the AUC for both of these models was numerically larger than the one that included APOE (after demographics) (0.76 for cognitive and 0.74 for MRI, versus 0.70 for APOE), the AUC for the models with either the cognitive or the MRI measures included (after demographics) did not differ significantly from the model with APOE (and demographics) at 5 years post-baseline (P = 0.07 and 0.15, respectively), although at 7 and 10 years the difference was significant (P = 0.04 and P = 0.01, respectively).
Discussion
While a number of studies have examined the risk of progression from normal cognition to MCI at the group level (Csernansky et al., 2005; Moghekar et al., 2013; Pettigrew et al., 2013; Roe et al., 2013; Toledo et al., 2014; Soldan et al., 2015; Vos et al., 2016), little is known about whether these same measures are useful for predicting progression at the individual level. These results demonstrate, for the first time to our knowledge, that biomarkers obtained when individuals are cognitively normal can be used to predict which individuals will develop clinical symptoms at 5, 7 or 10 years post-baseline. Both of the primary models examined (i.e. the Full Model and the Efficient Model) had sensitivities and specificities that approached or exceeded 0.80, the level recommended by biomarker workgroups as providing meaningful prediction (Ronald and Nancy Reagan Institute of the Alzheimer’s Association and National Institute on Aging Working Group on Biological Markers of Alzheimer’s Disease, 1998). The finding that 35% of false positive classifications by the Efficient Model at 5 years pertained to individuals who progressed at a later time point suggests that this model is quite sensitive in detecting the presence of preclinical Alzheimer’s disease. Moreover, the false negative rate was quite low (<2%).
Moreover, at 5 years post-baseline, each domain in both the Full Model and the Efficient Model significantly improved the accuracy of prediction when added consecutively to one another, demonstrating that each set of measures provided valuable, non-redundant, information with respect to the outcome. In contrast, the accuracy of prediction was slightly lower at 7 and 10 years post-baseline; this may suggest that the neurobiological changes associated with the development of Alzheimer’s disease are less pronounced 7 and 10 years prior to symptom onset, and thus less well captured by the measurements included in the study.
The incremental benefit in prediction that is generated by adding each of the domains consecutively to one another provides information that might be particularly useful for designing a screening strategy for a clinical trial. These analyses demonstrated several findings of note. First, age and education alone, combined with an individual’s APOE4 status and scores on two cognitive tests, was highly informative (sensitivity = 0.71, specificity = 0.71, AUC = 0.78). This suggests that it might be possible to enrich a sample of cognitively normal individuals likely to progress with these relatively non-invasive and inexpensive procedures.
Second, when APOE4 status was entered first in the model, the measure of CSF amyloid-β was not significant (with the same being true when CSF amyloid-β was added first, followed by APOE4). These findings likely reflect the strong association between APOE4 genotype and amyloid accumulation in the brain, as reflected in both in vivo (Morris et al., 2010; Resnick et al., 2015) and neuropathology studies (Gomez-Isla et al., 1996; Kok et al., 2009). Of note, information about an individual’s degree of amyloid accumulation and APOE4 status (adjusted by age and education) was slightly less accurate in predicting an individual’s outcome at 5 years compared to the model including APOE4 status and the two cognitive test scores, adjusted by demographics (sensitivity = 0.62, specificity = 0.70, AUC = 0.72).
Third, the addition of the MRI measures and CSF p-tau added relatively little in predictive power, above the other measures. Thus, it might be possible to forgo these expensive procedures, depending on the nature of the clinical trial that is being planned, although it is possible that alternative MRI measures (e.g. using 3 T MRI or different volumetric measures) would add more predictive power. It is, however, noteworthy that when CSF p-tau was added at the last step in the models, it significantly improved prediction, even though the sensitivity and specificity of the models were already quite good. Recent findings have demonstrated a moderate correlation between CSF p-tau and tau accumulation in the temporal lobe, as measured by tau PET imaging among cognitively normal individuals (using AV-1451) (Chhatwal et al., 2016; cf., Gordon et al., 2016), and elevated neocortical tau, particularly in the inferior temporal lobe, has been reported in patients with MCI (Johnson et al., 2016). Taken together with the results of the present study, these findings raise the possibility that the inclusion of tau imaging for screening subjects in a clinical trial of cognitively normal individuals may be highly informative.
Finally, it is also important to acknowledge the utility of the demographic variables in predicting the outcome. These analyses demonstrated that the prediction for an individual, based on demographics alone, yielded an AUC of 0.68 at all durations of follow-up (Table 4). This moderate predictability exemplifies the well-known increase in Alzheimer’s disease prevalence with age (Brookmeyer et al., 1998), which is also reflected by the fact that those who progressed to MCI in the present study were significantly older at baseline than those who remained cognitively normal (Table 2).
Our results complement studies that have examined different cognitive measures and Alzheimer’s disease biomarkers in relation to the onset of dementia among non-demented individuals (Amieva et al., 2004; Coupé et al., 2015; Stephan et al., 2015; Ritchie et al., 2016), which represents a later phase in the disease. For example, Coupé et al. (2015) reported that MRI biomarkers had moderate predictive utility on their own (AUC 0.64–0.73), but may have limited additional predictive power after accounting for demographics, cognitive status, and APOE4 genotype, at least on the group level (Stephan et al., 2015).
Taken as a whole, these analyses provide valuable information for researchers seeking to determine optimal methods for screening subjects for inclusion in clinical trials aimed at those with preclinical Alzheimer’s disease. The importance of selecting subjects likely to progress over the duration of a clinical trial is emphasized by a recent analysis of placebo data from MCI clinical trials; this report found that many subjects enrolled in these trials had limited progression over time, making it difficult to determine whether the subjects treated with active medication were, in fact, benefiting from treatment (Petersen et al., 2017). The findings reported here may, therefore, provide valuable information about how to select participants likely to progress for clinical trials in preclinical Alzheimer’s disease.
Limitations
This study has several limitations. It is important to acknowledge that although CSF amyloid-β correlates moderately with PET amyloid levels (Fagan et al., 2006; Vlassenko et al., 2016; Vos et al., 2016), the two are not identical; thus, results may differ if amyloid levels are measured with PET instead of CSF. Likewise, measures of CSF p-tau do not provide information about the regional distribution of tau throughout the brain and tau imaging may therefore provide additional valuable information that can be used in subject selection. Additionally, the use of different MRI measures (i.e. 3 T MRI or other volumetric measures) would likely give different results. The BIOCARD participants were well educated, primarily Caucasian, and the majority had a family history of dementia, so the results may not generalize to the population at large. Participants were also primarily middle-aged when first enrolled, thus the findings may not generalize to older cohorts. Additionally, the relatively small sample size did not allow us to test the reproducibility of these results, and prior work has shown that the specific measures selected may affect diagnostic accuracy (Frisoni et al., 2013). Future studies are therefore necessary to determine if similar findings would be obtained using more diverse groups of older individuals, and different biomarker measures (e.g. 3 T MRI scans; PET imaging for amyloid and tau).
Conclusion
In summary, these results indicate that it is feasible to predict on an individual basis which cognitively normal individuals are likely to progress to MCI at 5, 7 and 10 years post-baseline. This should facilitate the design of intervention studies aimed at the preclinical phase of Alzheimer’s disease, when treatments might be most effective.
Supplementary Material
Acknowledgements
We are grateful for the support of the entire BIOCARD study team at Johns Hopkins University and to the dedicated participants who continue to participate in the study. Additionally, we acknowledge the contributions of the Geriatric Psychiatry Branch of the intramural program of the NIMH who initiated the study (PI: Dr. Trey Sunderland).
Funding
This study is supported in part by grants from the National Institutes of Health: U19-AG03365, P50- AG005146 and P41-RR015241.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- AIC
Akaike Information Criterion
- AUC
area under the curve
- MCI
mild cognitive impairment
- p-tau
phosphorylated tau
- ROC
receiver operating characteristic
References
- Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr 1974; 19: 716–23. [Google Scholar]
- Albert M, Soldan A, Gottesman R, McKhann G, Sacktor N, Farrington L, et al. Cognitive changes preceding clinical symptom onset of mild cognitive impairment and relationship to ApoE genotype. Curr Alzheimer Res 2014; 11: 773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman H, Fox N, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva H, Letenneur L, Dartigues JF, Rouch-Leroyer I, Sourgen C, D’Alchée-Birée F, et al. Annual rate and predictors of conversion to dementia in subjects presenting mild cognitive impairment criteria defined according to a population-based study. Dement Geriart Cogn Disord 2004; 18: 87–93. [DOI] [PubMed] [Google Scholar]
- Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med 2013; 32: 5381–97. [DOI] [PubMed] [Google Scholar]
- Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health 1998; 88: 1337–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal J, Schultz AP, Marshall GA, Boot B, Gomez-Isla T, Dumurgier J, et al. Temporal T807 binding correlates with CSF tau and phosphor-tau in normal elderly. Neurology 2016; 87: 920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupé P, Fonov VS, Bernard C, Zandifar A, Eskildsen SF, Helmer C, et al. Detection of Alzheimer’s disease signature in MR images seven years before conversion to dementia: toward an early individual prognosis. Hum Brain Map 2015; 36: 4758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csernansky JG, Wang L, Swank J, Miller JP, Gado M, McKeel D, et al. Preclinical detection of Alzheimer’s disease: hippocampal shape and volume predict dementia onset in the elderly. Neuroimage 2005; 25: 783–92. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol 2006; 59: 512–19. [DOI] [PubMed] [Google Scholar]
- Fleisher A, Chen K, Quiroz Y, Jakimovich L, Gomez M, Langois C, et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant alzheimer disease kindred: A cross-sectional study. JAMA Neurol 2015; 72: 316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisoni GB, Bocchetta M, Chételat G, Rabinovici GD, de Leon MJ, Kaye J, et al. Imaging markers for Alzheimer disease: which vs. how. Neurology 2013; 81: 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, et al. Clinical and pathological correlates of Apolopoprotein E ɛ4 in Alzheimer’s disease. Ann Neurol 1996; 39: 62–70. [DOI] [PubMed] [Google Scholar]
- Gordon BA, Friedrichsen K, Brier M, Blazey T, Su Y, Christensen J, et al. The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain 2016; 139: 2249–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbe JM. Negative binomial regression. Cambridge, UK: Cambridge University Press; 2011. [Google Scholar]
- Holtzman DM. CSF biomarkers for Alzheimer's disease: current utility and potential future use. Neurobiol Aging 2011; 32 (Suppl 1): S4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K, Schultz A, Betensky R, Becker A, Sepulcre J, Rentz D, et al. Tau positron emission tomographic imaging in aging and early Alzheimer’s disease. Ann Neurol 2016; 79: 110–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 2009; 65: 650–7. [DOI] [PubMed] [Google Scholar]
- Lasser R, Ostrowitzki S, Scheltens P, Boada M, Dubois B, Dorflinger E, et al. Efficacy and safety of gantenerumab form the phase 3 scarlet road trial, a study of gantenerumab in patients with prodromal AD. J Prev Alzheimers Dis 2015; 2: 275. [Google Scholar]
- McIntosh MW, Pepe MS. Combining several screening tests: optimality of the risk score. Biometrics 2002; 58: 657–64. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hayman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MI, Younes L, Ratnanather JT, Brown T, Trinh H, Postell E, et al. The diffeomorphometry of temporal lobe structures in preclinical Alzheimer’s disease. Neuroimage Clin 2013; 3: 352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghekar A, Li S, Lu Y, Li M, Wang MC, Albert M, et al. Cerebrospinal fluid biomarker changes precede symptom onset of mild cognitive impairment. Neurology 2013; 12: 1753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts Aβ but not tau Alzheimer’s pathology in cognitively normal aging. Ann Neurol 2010; 67: 122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder K, Snider B, Milles S, Buckles V, Santacruz A, Bateman R, et al. Dominantly inherited Alzheimer network: facilitating research and clinical trials. Alzheimer Res Ther 2013; 5: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew C, Soldan A, Li S, Lu Y, Wang MC, Selnes OA, et al. Relationship of cognitive reserve and APOE status to the emergence of clinical symptoms in preclinical Alzheimers disease. Cogn Neurosci 2013; 4: 136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Thomas RG, Aisen PS, Mohs RC, Carrillo MC, Albert MS. Randomized clinical trials in mild cognitive impairment: sources of variability. Neurology 2017; 88: 1751–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman E, Langbaum J, Fleisher A, Caselli R, Chen K, Ayutyamont N, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis 2011; 26 (Suppl 3) 321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick S, Bilgel M, Moghekar A, An Y, Cai Q, Wang MC, et al. Changes in Abeta biomarkers and associations with ApoE genotype in 2 longitudinal cohorts. Neurobiol Aging 2015; 36: 2333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Carrière I, Berr C, Amieva H, Dartigues JF, Ancelin ML, et al. The clinical picture of Alzheimer’s disease in the decade before diagnosis: clinical and biomarker trajectories. J Clin Psychiatry 2016; 77: e305–11. [DOI] [PubMed] [Google Scholar]
- Roe CM, Fagan AM, Grant EA, Hassenstab J, Moulder KL, Dreyfus DM, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology 2013; 80: 1784–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald and Nancy Reagan Institute of the Alzheimer’s Association and National Institute on Aging Working Group on Biological Markers of Alzheimer’s Disease. Consensus Report of the Working Group on Molecular and Biochemical Markers of Alzheimer’s Disease. Ronald and Nancy Reagan Institute of the Alzheimer’s Association and National Institute on Aging Working Group on Biological Markers of Alzheimer’s Disease. Neurobiol Aging 1998; 19: 109–16. [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Williams L, Chen T, Ling Y, O’Gorman J, et al. Randomized placebo-controlled phase 1B study of the anti-beta amyloid antibody Aducanumab (BIIB037) in patients with prodromal or mild Alzheimer’s disease: interim results. J Prev Alzheimers Dis 2015; 2: 283. [Google Scholar]
- Soldan A, Pettigrew C, Lu Y, Wang M-C, Selnes O, Albert A, et al. Relationship of medial temporal lobe atrophy, APOE genotype, and cognitive reserve in preclinical AD. Hum Brain Map 2015; 36: 2826–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett D, Craft S, Fagan A, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011; 7: 280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon D, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014; 6: 228fs13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan BCM, Tzourio C, Auriacombe S, Amieva H, Dufouil C, Alpérovitch A, et al. Usefulness of data from magnetic resonance imaging to improve prediction of dementia: Population based cohort study. BMJ 2015; 350: h2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo JB, Weiner MC, Wolk DA, Da X, Chen K, Arnold SE, et al. Neuronal injury biomarkers and prognosis in ADNI subjects with normal cognition. Acta Neuropathol Commun 2014; 2: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, McCue L, Jasielec MS, Su Y, Gordon BA, Xiong C, et al. Imaging and cerebrospinal fluid biomarkers in early preclinical Alzheimer disease. Ann Neurol 2016; 80: 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJB, Gordon BA, Su Y, Visser PJ, Holtzman DM, Morris JC, et al. NIA-AA staging of preclinical Alzheimer disease: discordance and concordance of CSF and imaging biomarkers. Neurobiol Aging 2016; 44: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youden WJ. Index for rating diagnostic tests. Cancer 1950; 3: 32–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.