Total Synthesis of Annonaceous Acetogenins Belonging to the Non-Adjacent Bis-THF and Non-Adjacent THF-THP Sub-Classes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction





2. Total Syntheses of Non-Adjacent Bis-THF Acetogenins

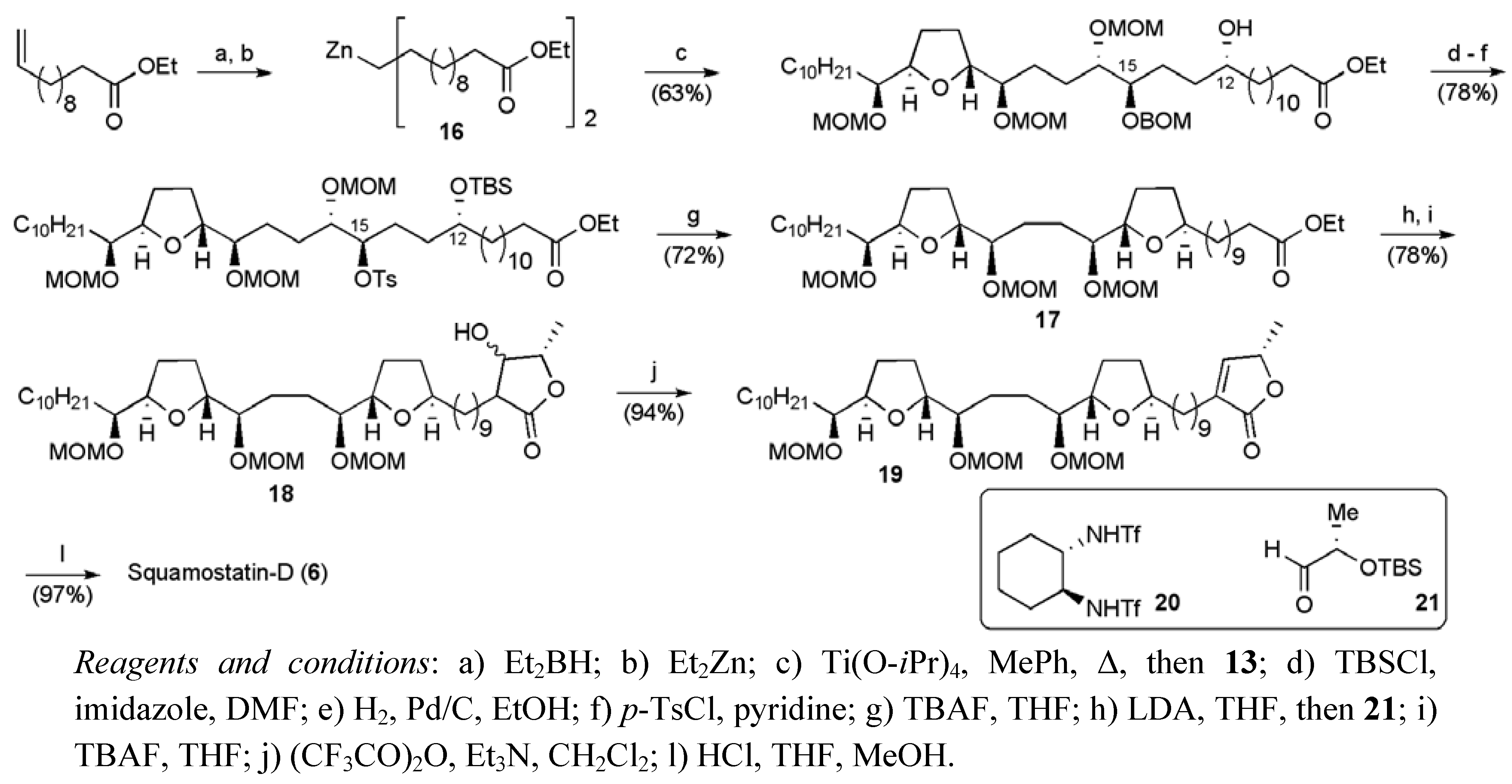
2.1. Total Synthesis of Squamostatin-D (6)



2.2. Total Synthesis of 4-Deoxygigantecin (2)




2.3. Total Syntheses of Gigantecin (1)










2.4. Total Synthesis of Squamostatin-C (Bullatanocin) (5)





2.5. Total Syntheses of cis-Sylvaticin (3)










2.6. Total Synthesis of Sylvaticin (7)


3. Total Synthesis of Non-Adjacent THF-THP Acetogenins

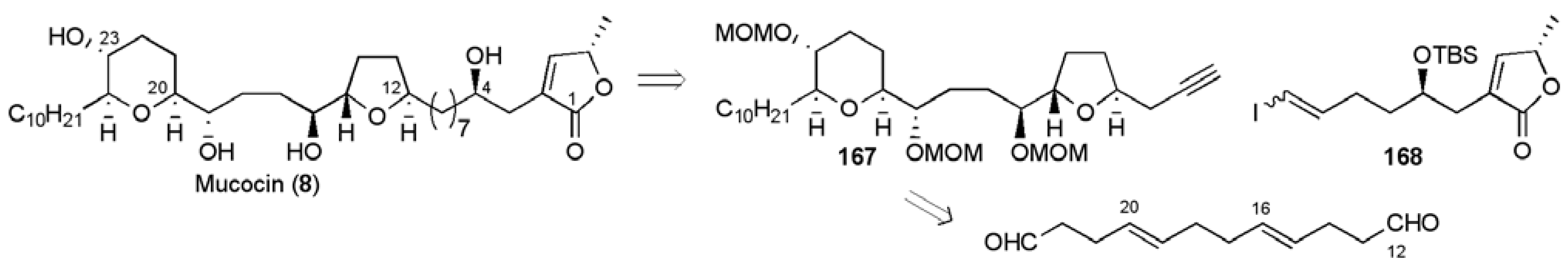
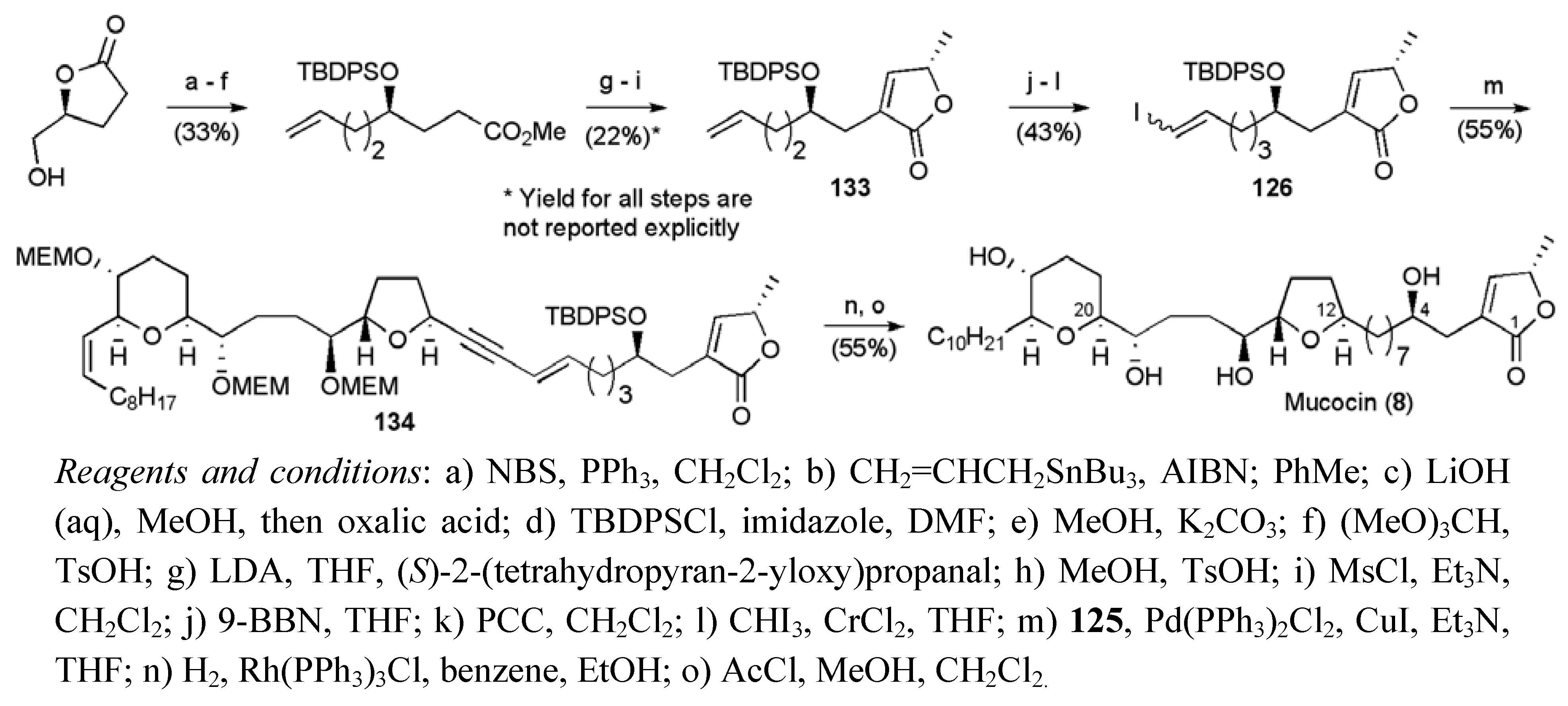
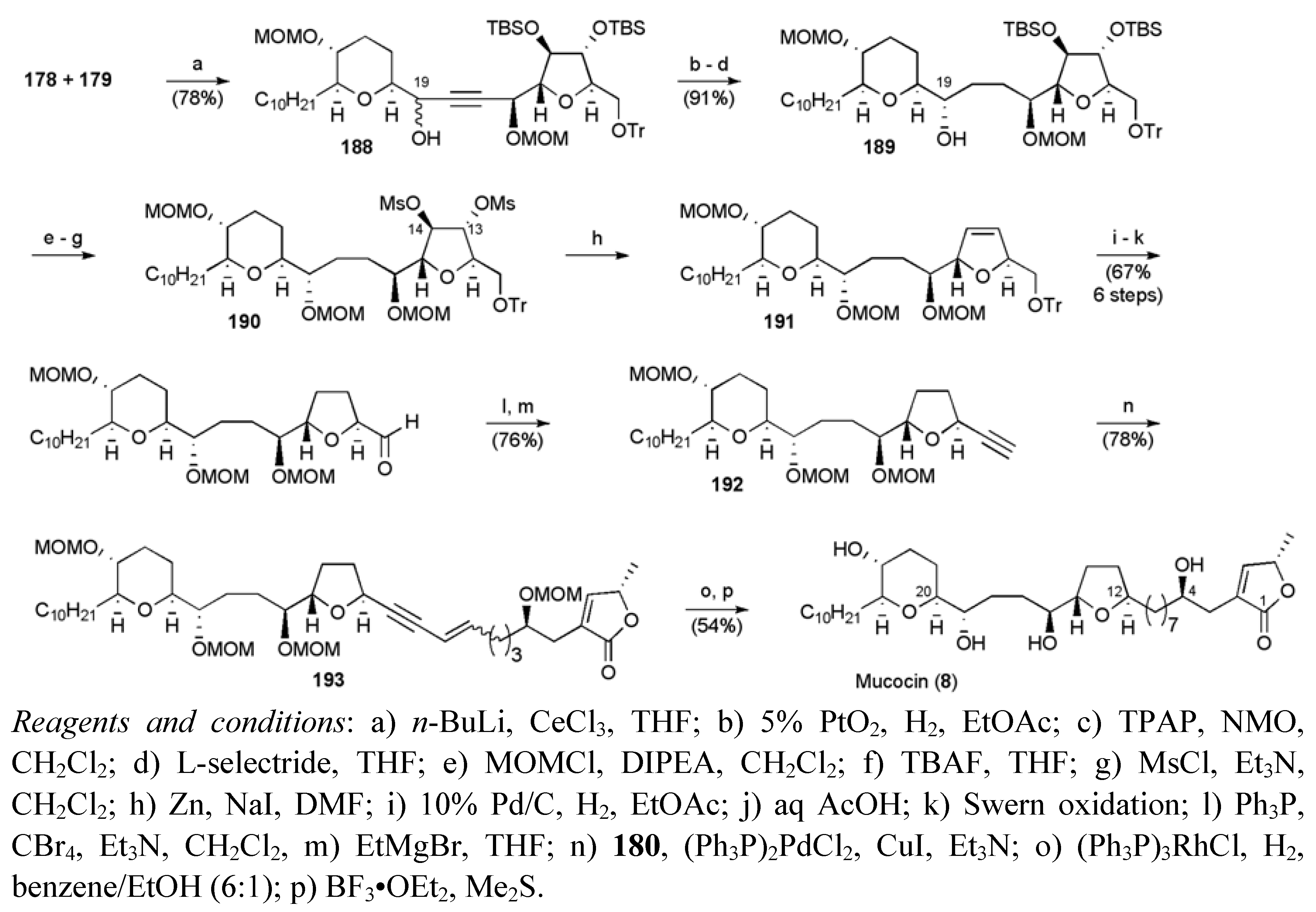
3.1. Total Syntheses of Mucocin (8)



























4. Summary
Acknowledgements
- Sample Availability: No samples are available from the authors.
References
- Alali, F.Q.; Liu, X.X.; McLaughlin, J.L. Annonaceous acetogenins: Recent progress. J. Nat. Prod. 1999, 62, 504–540. [Google Scholar] [CrossRef]
- Bermejo, A.; Figadere, B.; Zafra-Polo, M.C.; Barrachina, I.; Estornell, E.; Cortes, D. Acetogenins from annonaceae: Recent progress in isolation, synthesis and mechanisms of action. Nat. Prod. Rep. 2005, 22, 269–303. [Google Scholar] [CrossRef]
- Cavé, A.; Figadere, B.; Laurens, A.; Cortes, D. Acetogenins from Annonaceae. In Progress in the Chemistry of Organic Natural Products; Herz, W., Kirby, G.W., Moore, R.E., Steglisg, W., Tamm, C., Eds.; Springer-Verlag: New York, NY, USA, 1997; Volume 70, pp. 81–287. [Google Scholar]
- McLaughlin, J.L. Paw paw and cancer: Annonaceous acetogenins from discovery to commercial products. J. Nat. Prod. 2008, 71, 1311–1321. [Google Scholar] [CrossRef]
- Rupprecht, J.K.; Hui, Y.H.; McLaughlin, J.L. Annonaceous acetogenins - a review. J. Nat. Prod. 1990, 53, 237–278. [Google Scholar] [CrossRef]
- Zafra-Polo, M.C.; Figadere, B.; Gallardo, T.; Tormo, J.R.; Cortes, D. Natural acetogenins from annonaceae, synthesis and mechanisms of action. Phytochemistry 1998, 48, 1087–1117. [Google Scholar] [CrossRef]
- Zafra-Polo, M.C.; Gonzalez, M.C.; Estornell, E.; Sahpaz, S.; Cortes, D. Acetogenins from annonaceae, inhibitors of mitochondrial complex I. Phytochemistry 1996, 42, 253–271. [Google Scholar]
- Zeng, L.; Ye, Q.; Oberlies, N.H.; Shi, G.; Gu, Z.M.; He, K.; McLaughlin, J.L. Recent advances in annonaceous acetogenins. Nat. Prod. Rep. 1996, 13, 275–306. [Google Scholar] [CrossRef]
- Fang, X.P.; Rieser, M.J.; Gu, Z.M.; Zhao, G.X.; McLaughlin, J.L. Annonaceous acetogenins - an updated review. Phytochem. Anal. 1993, 4, 27–48. [Google Scholar] [CrossRef]
- Yu, J.G.; Hu, X.F.E.; Ho, D.K.; Bean, M.F.; Stephens, R.E.; Cassady, J.M.; Brinen, L.S. Clardy, J. Absolute stereochemistry of (+)-gigantecin from annona-coriacea (Annonaceae). J. Org. Chem. 1994, 59, 1598–1599. [Google Scholar]
- Curran, D.P.; Zhang, Q.S.; Lu, H.J. Gudipati, V. On the proof and disproof of natural product stereostructures: Characterization and analysis of a twenty-eight member stereoisomer library of murisolins and their Mosher ester derivatives. J. Am. Chem. Soc. 2006, 128, 9943–9956. [Google Scholar]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Prot. 2007, 2, 2451–2458. [Google Scholar] [CrossRef]
- Shi, G.E.; Zeng, L.; Gu, Z.M.; Macdougal, J.M.; Mclaughlin, J.L. Absolute stereochemistries of sylvaticin and 12,15-cis-sylvaticin, bioactive C-20,23-cis non-adjacent bistetrahydrofuran annonaceous acetogenins, from rollinia-mucosa. Heterocycles 1995, 41, 1785–1796. [Google Scholar] [CrossRef]
- Born, L.; Lieb, F.; Lorentzen, J.P.; Moeschler, H.; Nonfon, M.; Sollner, R.; Wendisch, D. The relative configuration of acetogenins isolated from annona-squamosa - annonin-I (Squamocin) and annonin-VI. Planta Med. 1990, 56, 312–316. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Murasaki, C.; Shimada, H.; Nishioka, S.; Kakinuma, K.; Singh, S.; Singh, M.; Gupta, Y.K.; Sahai, M. Annonaceous Acetogenins from the seeds of annona-squamosa - non-adjacent bis-tetrahydrofuranic acetogenins. Chem. Pharm. Bull. 1994, 42, 1175–1184. [Google Scholar] [CrossRef]
- Hoye, T.R.; Suhadolnik, J.C. On the Stereochemistry of the bistetrahydrofuranyl moiety of uvaricin - proton chemical-shifts can play a crucial role in complex structure determination. J. Am. Chem. Soc. 1987, 109, 4402–4403. [Google Scholar] [CrossRef]
- Hoye, T.R.; Zhuang, Z.P. Validation of the H-1-NMR Chemical-shift method for determination of stereochemistry in the bis(tetrahydrofuranyl) moiety of uvaricin-related acetogenins from annonaceae - rolliniastatin-1 (and asimicin). J. Org. Chem. 1988, 53, 5578–5580. [Google Scholar] [CrossRef]
- Ghani, S.B.A.; Chapman, J.M.; Figadere, B.; Herniman, J.M.; Langley, G.J.; Niemann, S.; Brown, R.C.D. Total synthesis and stereochemical assignment of cis-uvariamicin I and cis-reticulatacin. J. Org. Chem. 2009, 74, 981–988. [Google Scholar]
- Hu, Y.L.; Cecil, A.R.L.; Frank, X.; Gleye, C.; Figadere, B.; Brown, R.C.D. Natural cis-solamin is a mixture of two tetra-epimeric diastereoisomers: Biosynthetic implications for annonaceous acetogenins. Org. Biomol. Chem. 2006, 4, 1217–1219. [Google Scholar] [CrossRef]
- Zhao, G.X.; Hui, Y.H.; Rupprecht, J.K.; Mclaughlin, J.L.; Wood, K.V. Additional bioactive compounds and trilobacin, a novel highly cytotoxic acetogenin, from the bark of asimina-triloba. J. Nat. Prod. 1992, 55, 347–356. [Google Scholar] [CrossRef]
- Zhao, G.X.; Miesbauer, L.R.; Smith, D.L.; Mclaughlin, J.L. Asimin, asiminacin, and asiminecin - novel highly cytotoxic asimicin isomers from asimina-triloba. J. Med. Chem. 1994, 37, 1971–1976. [Google Scholar] [CrossRef]
- Oberlies, N.H.; Chang, C.J.; McLaughlin, J.L. Structure-activity relationships of diverse annonaceous acetogenins against multidrug resistant human mammary adenocarcinoma (MCF-7/Adr) cells. J. Med. Chem. 1997, 40, 2102–2106. [Google Scholar] [CrossRef]
- Oberlies, N.H.; Croy, V.L.; Harrison, M.L.; McLaughlin, J.L. The annonaceous acetogenin bullatacin is cytotoxic against multidrug-resistant human mammary adenocarcinoma cells. Cancer Lett. 1997, 115, 73–79. [Google Scholar] [CrossRef]
- Ahammadsahib, K.I.; Hollingworth, R.M.; McGovren, J.P.; Hui, Y.H.; McLaughlin, J.L. Mode of action of bullatacin: A potent antitumor and pesticidal annonaceous acetogenin. Life Sci. 1993, 53, 1113–1120. [Google Scholar] [CrossRef]
- Degli Esposti, M.; Ghelli, A.; Ratta, M.; Cortes, D.; Estornell, E. Natural substances (Acetogenins) from the family annonanaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (complex I). Biochem. J. 1994, 301, 161–167. [Google Scholar]
- Londershausen, M.; Leicht, W.; Lieb, F.; Moeschler, H.; Weiss, H. Molecular mode of action of annonins. Pestic.Sci. 1991, 33, 427–438. [Google Scholar] [CrossRef]
- Morré, D.J.; de Cabo, R.; Farley, C.; Oberlies, N.H.; McLaughlin, J.L. Mode of action of bullatacin, a potent antitumor acetogenin: Inhibition of NADH oxidase activity. Life Sci. 1995, 56, 343–348. [Google Scholar]
- Abe, M.; Kubo, A.; Yamamoto, S.; Hatoh, Y.; Murai, M.; Hattori, Y.; Makabe, H.; Nishioka, T.; Miyoshi, H. Dynamic function of the spacer region of acetogenins in the inhibition of bovine mitochondrial NADH-ubiquinone oxidoreductase (complex I). Biochemistry 2008, 47, 6260–6266. [Google Scholar]
- González, M.C.; Lavaud, C.; Gallardo, T.; Zafra-Polo, M.C.; Cortes, D. New method for the determination of the absolute stereochemistry in antitumoral annonaceous acetogenins. Tetrahedron 1998, 54, 6079–6088. [Google Scholar]
- Motoyama, T.; Yabunaka, H.; Miyoshi, H. Essential structural factors of acetogenins, potent inhibitors of mitochondrial complex I. Bioorg. Med. Chem. Lett. 2002, 12, 2089–2092. [Google Scholar] [CrossRef]
- Tormo, J.R.; Estornell, E.; Gallardo, T.; González, M.C.; Cáve, A.; Granell, S.; Cortes, D.; Zafra-Polo, M.C. Gamma-lactone functionalised antitumoral acetogenins are the most potent inhibitors of mitochondrial complex I. Bioorg. Med. Chem. Lett. 2001, 11, 681–684. [Google Scholar]
- Marshall, J.A.; Hinkle, K.W. Hagedorn, C.E. Recent Developments in the synthesis of annonaceous acetogenins. Isr. J. Chem. 1997, 37, 97–107. [Google Scholar]
- Figadere, B.; Cave, A. Total Stereoselective Synthesis of Acetogenins of Annonaceae: A New Class of Bioactive Polyketides. In Studies in Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier Science: Amsterdam, The Netherlands, 1996; Volume 18, pp. 193–227. [Google Scholar]
- Figadere, B. Syntheses of acetogenins of annonaceae: A new class of bioactive polyketides. Acc. Chem. Res. 1995, 28, 359–365. [Google Scholar] [CrossRef]
- Casiraghi, G.; Zanardi, F.; Battistini, L.; Rassu, G.; Appendino, G. Current Advances in the chemical synthesis of annonaceous acetogenins and relatives. Chemtracts Org. Chem. 1998, 11, 803–827. [Google Scholar]
- Hoppe, R.; Scharf, H.D. Annonaceous acetogenins - synthetic approaches towards a novel class of natural products. Synthesis 1995, 1447–1464. [Google Scholar] [CrossRef]
- Li, N.; Shi, Z.; Tang, Y.; Chen, J.; Li, X. Recent progress on the total synthesis of acetogenins from annonaceae. Beilstein J. Org. Chem. 2008, 4, No. 48. [Google Scholar]
- Marshall, J.A.; Jiang, H.J. Total synthesis of the non-adjacent bis-tetrahydrofuran annonaceous acetogenin squamostatin-D. J. Org. Chem. 1998, 63, 7066–7071. [Google Scholar] [CrossRef]
- Marshall, J.A.; Hinkle, K.W. Synthesis of anti-homoallylic alcohols and monoprotected 1,2-diols through InCl3-promoted addition of allylic stannanes to aldehydes. J. Org. Chem. 1995, 60, 1920–1921. [Google Scholar] [CrossRef]
- Yao, Z.J.; Wu, Y.L. Total synthesis of (10-ξ,15R,16S,19S,20S,34R)-corossoline. Tetrahedron Lett. 1994, 35, 157–160. [Google Scholar] [CrossRef]
- Fang, X.P.; Anderson, J.E.; Smith, D.L.; Wood, K.V.; McLaughlin, J.L. Giganenin, a highly potent monotetrahydrofuran acetogenin and 4-deoxygigantecin from goniothalamus-giganteus. Heterocycles 1992, 34, 1075–1083. [Google Scholar] [CrossRef]
- Makabe, H. Synthesis of annonaceous acetogenins from muricatacin. Biosci. Biotech. Biochem. 2007, 71, 2367–2374. [Google Scholar] [CrossRef]
- Makabe, H.; Tanaka, A.; Oritani, T. Total synthesis of solamin and reticulatacin. J. Chem. Soc. Perkin Trans. I 1994, 1975–1981. [Google Scholar]
- Makabe, H.; Tanaka, A.; Oritani, T. Total synthesis of (+)-4-deoxygigantecin. Tetrahedron 1998, 54, 6329–6340. [Google Scholar] [CrossRef]
- Makabe, H.; Tanaka, A.; Oritani, T. Synthesis of (–)-muricatacin. Biosci. Biotech. Biochem. 1993, 57, 1028–1029. [Google Scholar] [CrossRef]
- White, J.D.; Somers, T.C.; Reddy, G.N. Degradation and absolute configurational assignment to C-34-botryococcene. J. Org. Chem. 1992, 57, 4991–4998. [Google Scholar] [CrossRef]
- Alkofahi, A.; Rupprecht, J.K.; Liu, Y.M.; Chang, C.J.; Smith, D.L.; McLaughlin, J.L. Gigantecin —A novel antimitotic and cytotoxic acetogenin, with nonadjacent tetrahydrofuran rings, from goniothalamus-giganteus (Annonaceae). Experientia 1990, 46, 539–541. [Google Scholar] [CrossRef]
- Crimmins, M.T.; She, J. Enantioselective total synthesis of (+)-gigantecin: Exploiting the asymmetric glycolate aldol reaction. J. Am. Chem. Soc. 2004, 126, 12790–12791. [Google Scholar] [CrossRef]
- Hoye, T.R.; Eklov, B.M.; Jeon, J. Khoroosi, M. Sequencing of three-component olefin metatheses: total synthesis of either (+)-gigantecin or (+)-14-deoxy-9-oxygigantecin. Org. Lett. 2006, 8, 3383–3386. [Google Scholar]
- Crimmins, M.T.; She, J. An improved procedure for asymmetric aldol additions with N-acyl oxazolidinones, oxazolidinethiones and thiazolidinethiones. Synlett 2004, 1371–1374. [Google Scholar]
- Crimmins, M.T.; Emmitte, K.A.; Katz, J.D. Diastereoselective alkylations of oxazolidinone glycolates: A useful extension of the evans asymmetric alkylation. Org. Lett. 2000, 2, 2165–2167. [Google Scholar] [CrossRef]
- Hoye, T.R.; Ye, Z.X. Highly efficient synthesis of the potent antitumor annonaceous acetogenin (+)-parviflorin. J. Am. Chem. Soc. 1996, 118, 1801–1802. [Google Scholar] [CrossRef]
- Gu, Z.M.; Fang, X.P.; Rieser, M.J.; Hui, Y.H.; Miesbauer, L.R.; Smith, D.L.; Wood, K.V.; McLaughlin, J.L. New cytotoxic annonaceous acetogenins - bullatanocin and cis-bullatanocinone and trans-bullatanocinone, from annona-bullata (Annonaceae). Tetrahedron 1993, 49, 747–754. [Google Scholar]
- Gu, Z.M.; Zeng, L.; Fang, X.P.; Colmansaizarbitoria, T.; Huo, M.; McLaughlin, J.L. Determining absolute-configurations of stereocenters in annonaceous acetogenins through formaldehyde acetal derivatives and Mosher ester methodology. J. Org. Chem. 1994, 59, 5162–5172. [Google Scholar]
- Shimada, H.; Nishioka, S.; Singh, S.; Sahai, M.; Fujimoto, Y. Absolute stereochemistry of non-adjacent bis-tetrahydrofuranic acetogenins. Tetrahedron Lett. 1994, 35, 3961–3964. [Google Scholar] [CrossRef]
- Zhu, L.; Mootoo, D.R. Synthesis of nonadjacently linked tetrahydrofurans: An iodoetherification and olefin metathesis approach. Org. Lett. 2003, 5, 3475–3478. [Google Scholar] [CrossRef]
- Zhu, L.; Mootoo, D.R. Total synthesis of the nonadjacently linked bis-tetrahydrofuran acetogenin bullatanocin (squamostatin C). J. Org. Chem. 2004, 69, 3154–3157. [Google Scholar] [CrossRef]
- Dabideen, D.; Ruan, Z.M.; Mootoo, D.R. 1,2-O-Isopropylidene-5-alkene templates for the synthesis of oligo-tetrahydrofurans. Tetrahedron 2002, 58, 2077–2084. [Google Scholar] [CrossRef]
- Evans, P.A.; Murthy, V.S. Enantioselective construction of the tetrahydropyran and tetrahydrofuran fragments of the antitumor agent mucocin from a common intermediate. Tetrahedron Lett. 1999, 40, 1253–1256. [Google Scholar] [CrossRef]
- Hoye, T.R.; Mayer, M.J.; Vos, T.J.; Ye, Z.X. A general, Practical, and versatile strategy for accessing omega-functional 1,2-Diols of high enantiomeric excess. J. Org. Chem. 1998, 63, 8554–8557. [Google Scholar] [CrossRef]
- Sinha, S.C.; Sinha, A.; Sinha, S.C.; Keinan, E. Tandem oxidative cyclization with rhenium oxide. Total synthesis of 17,18-bisepi-goniocin. J. Am. Chem. Soc. 1997, 119, 12014–12015. [Google Scholar]
- Donohoe, T.J.; Harris, R.M.; Burrows, J.; Parker, J. Total synthesis of (+)-cis-sylvaticin: Double oxidative cyclization reactions catalyzed by osmium. J. Am. Chem. Soc. 2006, 128, 13704–13705. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Harris, R.M.; Williams, O.; Hargaden, G.C.; Burrows, J.; Parker, J. Concise syntheses of the natural products (+)-sylvaticin and (+)-cis-sylvaticin. J. Am. Chem. Soc. 2009, 131, 12854–12861. [Google Scholar]
- Bhunnoo, R.A.; Hobbs, H.; Laine, D.I.; Light, M.E.; Brown, R.C.D. Synthesis of the non-adjacent bis-thf core of cis-sylvaticin using a double oxidative cyclisation. Org. Biomol. Chem. 2009, 7, 1017–1024. [Google Scholar] [CrossRef]
- Brown, L.J.; Spurr, I.B.; Kemp, S.C.; Camp, N.P.; Gibson, K.R.; Brown, R.C.D. Total synthesis of cis-sylvaticin. Org. Lett. 2008, 10, 2489–2492. [Google Scholar]
- Piccialli, V. Oxidative Cyclization of dienes and polyenes mediated by transition-metal-oxo species. Synthesis 2007, 2585–2607. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Butterworth, S. Oxidative cyclization of diols derived from 1,5-dienes: Formation of enantiopure cis-tetrahydrofurans by using catalytic osmium tetroxide; formal synthesis of (+)-cis-solamin. Angew.Chem. Int. Ed. 2005, 44, 4766–4768. [Google Scholar] [CrossRef]
- Keum, G.; Hwang, C.H.; Kang, S.B.; Kim, Y.; Lee, E. Stereoselective Syntheses of rolliniastatin 1, rollimembrin, and membranacin. J. Am. Chem. Soc. 2005, 127, 10396–10399. [Google Scholar] [CrossRef]
- Ready, J.M.; Jacobsen, E.N. A Practical oligomeric [(Salen)Co] catalyst for asymmetric epoxide ring-opening reactions. Angew. Chem. Int. Ed. 2002, 41, 1374–1377. [Google Scholar] [CrossRef]
- Brown, R.C.D.; Bataille, C.J.; Hughes, R.M.; Kenney, A.; Luker, T.J. Permanganate oxidation of 1,5,9-trienes: Stereoselective synthesis of tetrahydrofuran-containing fragments. J. Org. Chem. 2002, 67, 8079–8085. [Google Scholar] [CrossRef]
- Hu, Y.L.; Brown, R.C.D. A Metal-oxo mediated approach to the synthesis of 21,22-diepi-membrarollin. Chem. Commun. 2005, 5636–5637. [Google Scholar]
- Morris, C.L.; Hu, Y.L.; Head, G.D.; Brown, L.J.; Whittingham, W.G.; Brown, R.C.D. Oxidative cyclization reactions of trienes and dienynes: Total synthesis of membrarollin. J. Org. Chem. 2009, 74, 981–988. [Google Scholar]
- Cecil, A.R.L.; Hu, Y.L.; Vicent, M.J.; Duncan, R.; Brown, R.C.D. Total synthesis and preliminary biological evaluation of cis-solamin isomers. J. Org. Chem. 2004, 69, 3368–3374. [Google Scholar]
- Cepleanu, F.; Ohtani, K.; Hamburger, M.; Gupta, M.P.; Solis, P. Hostettmann, K. Novel acetogenins from the leaves of annona-purpurea. Helv. Chim. Acta 1993, 76, 1379–1388. [Google Scholar]
- Mikolajczak, K.J.; Madrigal, R.V.; Rupprecht, J.K.; Hui, Y.H.; Liu, Y.M.; Smith, D.L.; Mclaughlin, J.L. Sylvaticin—A new cytotoxic and insecticidal acetogenin from rollinia-sylvatica (Annonaceae). Experientia 1990, 46, 324–327. [Google Scholar] [CrossRef]
- Donohoe, T.J.; Williams, O.; Churchill, G.H. Hydride shift generated oxonium ions: Evidence for mechanism and intramolecular trapping experiments to form trans-THF derivatives. Angew. Chem. Int. Ed. 2008, 47, 2869–2871. [Google Scholar] [CrossRef]
- Takahashi, S.; Hongo, Y.; Tsukagoshi, Y.; Koshino, H. Structural determination of montanacin D by total synthesis. Org. Lett. 2008, 10, 4223–4226. [Google Scholar] [CrossRef]
- Takahashi, S.; Takahashi, R.; Hongo, Y.; Koshino, H.; Yamaguchi, K.; Miyagi, T. Synthesis of all possible isomers corresponding to the proposed structure of montanacin E, and their antitumor activity. J. Org. Chem. 2009, 74, 6382–6385. [Google Scholar] [CrossRef]
- Shi, G.E.; Alfonso, D.; Fatope, M.O.; Zeng, L.; Gu, Z.M.; Zhao, G.X.; He, K.; Macdougal, J.M.; Mclaughlin, J.L. Mucocin—A new annonaceous acetogenin bearing a tetrahydropyran ring. J. Am. Chem. Soc. 1995, 117, 10409–10410. [Google Scholar]
- Baurle, S.; Hoppen, S.; Koert, U. Total synthesis of (–)-mucocin. Angew.Chem. Int. Ed. 1999, 38, 1263–1266. [Google Scholar] [CrossRef]
- Hoppen, S.; Baurle, S.; Koert, U. A convergent total synthesis of (–)-mucocin: An acetogenin from annonaceae. Chem. Eur. J. 2000, 6, 2382–2396. [Google Scholar] [CrossRef]
- Hoppen, S.; Bauerle, S.; Koert, U. A convergent total synthesis of (-)-mucocin: An acetogenin from annonaceae (vol 6, pg 2382, 2000). Chem. Eur. J. 2000, 6, 2906–2906. [Google Scholar]
- Nicolaou, K.C.; Prasad, C.V.C.; Somers, P.K.; Hwang, C.K. Activation of 6-Endo over 5-Exo hydroxy epoxide openings - stereoselective and ring selective synthesis of tetrahydrofuran and tetrahydropyran systems. J. Am. Chem. Soc. 1989, 111, 5330–5334. [Google Scholar] [CrossRef]
- Keinan, E.; Sinha, A.; Yazbak, A.; Sinha, S.C.; Sinha, S.C. Towards Chemical libraries of annonaceous acetogenins. Pure Appl. Chem. 1997, 423–430. [Google Scholar]
- Sinha, S.C.; Keinan, E. Total synthesis of (+)-aspicilin. The naked carbon skeleton strategy vs the bioorganic approach. J. Org. Chem. 1997, 62, 377–386. [Google Scholar] [CrossRef]
- Neogi, P.; Doundoulakis, T.; Yazbak, A.; Sinha, S.C.; Sinha, S.C.; Keinan, E. Total synthesis of mucocin. J. Am. Chem. Soc. 1998, 120, 11279–11284. [Google Scholar] [CrossRef]
- Evans, P.A.; Cui, J.; Gharpure, S.J.; Polosukhin, A.; Zhang, H.R. Enantioselective total synthesis of the potent antitumor agent (–)-mucocin using a temporary silicon-tethered ring-closing metathesis cross-coupling reaction. J. Am. Chem. Soc. 2003, 125, 14702–14703. [Google Scholar]
- Evans, P.A.; Murthy, V.S. Enantioselective synthesis of the 4-hydroxy buteneolide terminus of mucocin and related annonaceous acetogenins. Tetrahedron Lett. 1998, 39, 9627–9628. [Google Scholar]
- Evans, P.A.; Murthy, V.S. Enantioselective synthesis of the 4-hydroxy buteneolide terminus of mucocin and related annonaceous acetogenins (1998, vol. 39, p. 9627). Tetrahedron Lett. 1999, 40, 1423–1423. [Google Scholar]
- Evans, P.A.; Roseman, J.D. Stereoselective synthesis of the 2,6-disubstituted tetrahydropyran-3-ol of the potent antitumor agent mucocin via an acyl radical cyclization. Tetrahedron Lett. 1997, 38, 5249–5252. [Google Scholar] [CrossRef]
- Evans, P.A.; Cui, B.; Buffone, G.P. Diastereoselective temporary silicon-tethered ring-closing-metathesis reactions with prochiral alcohols: A new approach to long-range asymmetric induction. Angew. Chem. Int. Ed. 2003, 42, 1734–1737. [Google Scholar] [CrossRef]
- Inoki, S.; Mukaiyama, T. A convenient method for the stereoselective preparation of trans-2-hydroxymethyltetrahydrofurans by the oxidative cyclization of 5-hydroxy-1-alkenes with molecular-oxygen catalyzed by cobalt(II) complex. Chem. Lett. 1990, 67–70. [Google Scholar]
- Evans, P.A.; Cui, J.; Gharpure, S.J. Stereoselective construction of Cis-2,6-disubstituted tetrahydropyrans via the reductive etherification of δ-trialkylsilyloxy substituted ketones: Total synthesis of (–)-centrolobine. Org. Lett. 2003, 5, 3883–3885. [Google Scholar] [CrossRef]
- Evans, P.A.; Cui, J.; Gharpure, S.J.; Hinkle, R.J. Stereoselective construction of cyclic ethers using a tandem two-component etherification: Elucidation of the role of bismuth tribromide. J. Am. Chem. Soc. 2003, 125, 11456–11457. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Zhang, Y.; Diaz, F.A. Total synthesis of (–)-mucocin. Org. Lett. 2006, 8, 2369–2372. [Google Scholar] [CrossRef]
- Mori, K.; Otsuka, T. Pheromone synthesis. 59. synthesis of both the enantiomers of Erythro-6-acetoxy-5-hexadecanolide—The major component of a mosquito oviposition attractant pheromone. Tetrahedron 1983, 39, 3267–3269. [Google Scholar]
- Keinan, E.; Sinha, S.C.; Sinhabagchi, A.; Wang, Z.M.; Zhang, X.L.; Sharpless, K.B. Synthesis of all 4 isomers of disparlure using osmium-catalyzed asymmetric dihydroxylation. Tetrahedron Lett. 1992, 33, 6411–6414. [Google Scholar] [CrossRef]
- Hoye, T.R.; Jeffrey, C.S.; Tennakoon, M.A.; Wang, J.Z.; Zhao, H.Y. Relay ring-closing metathesis (RRCM): A Strategy for directing metal movement throughout olefin metathesis sequences. J. Am. Chem. Soc. 2004, 126, 10210–10211. [Google Scholar]
- Zhu, L.; Mootoo, D.R. Synthesis of the non-classical acetogenin mucocin: A Modular approach based on olefinic coupling reactions. Org. Biomol. Chem. 2005, 3, 2750–2754. [Google Scholar] [CrossRef]
- Takahashi, S.; Kubota, A.; Nakata, T. Stereoselective total synthesis of mucocin, an antitumor agent. Angew. Chem. Int. Ed. 2002, 41, 4751–4754. [Google Scholar] [CrossRef]
- Hori, N.; Matsukura, H.; Matsuo, G.; Nakata, T. Efficient strategy for the iterative synthesis of trans-fused polycyclic ethers via smi2-induced reductive intramolecular cyclization. Tetrahedron 2002, 58, 1853–1864. [Google Scholar] [CrossRef]
- Takahashi, S.; Nakata, T. Total synthesis of an anticancer agent, mucocin. 1. stereoselective synthesis of the left-half segment. Tetrahedron Lett. 1999, 40, 723–726. [Google Scholar]
- Takahashi, S.; Nakata, T. Total synthesis of an anticancer agent, mucocin. 2. A Novel approach to a gamma-hydroxy butenolide derivative and completion of total synthesis. Tetrahedron Lett. 1999, 40, 727–730. [Google Scholar]
- Takahashi, S.; Nakata, T. Total synthesis of an antitumor agent, mucocin, based on the "chiron approach". J. Org. Chem. 2002, 67, 5739–5752. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Spurr, I.B.; Brown, R.C.D. Total Synthesis of Annonaceous Acetogenins Belonging to the Non-Adjacent Bis-THF and Non-Adjacent THF-THP Sub-Classes. Molecules 2010, 15, 460-501. https://doi.org/10.3390/molecules15010460
Spurr IB, Brown RCD. Total Synthesis of Annonaceous Acetogenins Belonging to the Non-Adjacent Bis-THF and Non-Adjacent THF-THP Sub-Classes. Molecules. 2010; 15(1):460-501. https://doi.org/10.3390/molecules15010460
Chicago/Turabian StyleSpurr, Ian B., and Richard C. D. Brown. 2010. "Total Synthesis of Annonaceous Acetogenins Belonging to the Non-Adjacent Bis-THF and Non-Adjacent THF-THP Sub-Classes" Molecules 15, no. 1: 460-501. https://doi.org/10.3390/molecules15010460