
Preprint
Review
Future Perspectives on the Application of the Oxford Nanopore® MinION™ Sequencer in Cultural Heritage Biodeterioration Studies
Altmetrics
Downloads
252
Views
306
Comments
0



This version is not peer-reviewed
Abstract
The present work provides an updated overview of the application of the Oxford Nanopore® MinION™ sequencer in cultural heritage biodeterioration studies, while also providing a holistic discussion of possible future perspectives for their utilization in this research field. Due to the peculiar characteristics of this device, the last few years have seen the steady rise on the application of this system in a variety of cultural heritage materials, having been useful to understand microbial biodeteriogens diversity and some of their metabolic and biodeteriogenic features. Considering the immense potential for application of this system, this manuscript discusses further possibilities of the technique aiming to help understand critical questions on the cultural heritage biodeterioration area. Its application in various differential contexts, has opened the doors for their putative usage on other interesting sub-areas of research worthy of future investigations, including: biodeteriogens genome and transcriptome analysis, metatranscriptomics, biodeteriorative metabolism studies, inter and intra kingdom interactions analysis, resistome profiling, object history and context studies, in situ applications, bioprospecting and biotechnology.
Keywords:
Subject: Biology and Life Sciences - Life Sciences
1. Introduction
Humankind cultural heritage objects, relics and sites can be undesirably altered and seriously damaged from the growth and metabolic activities of living organisms [1,2,3]. These biodeterioration processes can occur at both indoor (e.g., museums) and outdoor environments (e.g., monuments), posing a serious risk for worldwide historical sites, properties and objects. Materials such as paper, ceramics, textiles, glass and stone, or objects including parchments, books, paintings, frescoes, vitrails, photographs, sculptures and funerary accessories, can be colonized and deteriorated through microorganisms’ aesthetic, mechanical, acid and enzymatic vital actions and manipulations [4]. Biodeterioration can thus be a result of the impact of various organisms (bacteria, cyanobacteria, microalgae, archaea, fungi and lichens [4]), and requires that protective measures are constantly considered, developed and implemented. Depending on specific conditions and the substrate type, some of these microorganisms can also contribute to the protection of the materials, thus displaying a biodeteriorative/bioprotective dualistic effect/nature [5,6]. For these reasons, molecular techniques, such as DNA sequencing, have been largely applied to investigate, understand and monitor biological colonization on art objects and cultural heritage monuments for more than two decades [7,8,9]. Justifiably, the current focus resides in the application of Next-Generation-Sequencing (NGS) methodologies, since they have powerfully expanded the possibility to characterize microbial communities in a cheaper, quick and holistic manner [10,11,12,13,14,15,16]. Due to the peculiar characteristics of this device, the last few years have seen the steady rise of the popular Oxford Nanopore® MinION™ sequencer in this research area, which has been applied in a variety of cultural heritage materials [8,9,17]. MinION™ is a small (Figure 1), mobile, relatively inexpensive, long read DNA/RNA sequencer, with an enormous range of applications. The technology relies on nanopores (protein pores), acting as biosensors to detect negatively charged single-stranded DNA or RNA molecules driven through the nanopore along an ionic current, and decoding of electric variations with computational algorithms [16]. In this review article, we aim to provide a brief update summary of previous studies using the MinION™ sequencer, while simultaneously providing a holistic discussion of possible future directions, additional applications and associated impacts of their utilization in the field of cultural heritage materials biodeterioration (in order to stimulate further discussions).
2. Application of Oxford Nanopore® in Cultural Heritage Biodeterioration Studies
So far, the Oxford Nanopore® MinION™ sequencer has been applied in a variety of cultural heritage materials including: stone monuments, granite chapels, salt-weathered buildings, petroglyph sites, oil paintings, drawings, textiles, waxes, bronze statues, waterlogged archeological wood pirogues, iron nails from a whale skeleton, documents and in museum environments (Table 1).
Grottoli and colleagues [18] used a gene sequencing tailored approach (ITS, 16S and 18S rDNA regions) to apply the Oxford Nanopore® MinION™ sequencer in a study in the hypogeum of Basilica di San Nicola, in Carcere Church (Rome, Italy). The authors developed and tested a bioinformatics approach, named “AmpLIcon SequencIng Analysis” (ALISIA) to evaluate their results, and verified a feeble overlap between this approach and culture dependent methods. Kisová and colleagues [19] used a metabarcoding approach (ITS, 16S and 28S rDNA regions) to study the microbiomes of funeral accessories. The authors were able to characterize biodeteriorative microorganisms, including bacteria responsible for metal corrosion and bio-mineralization, and entomopathogenic and phytopathogenic fungi problematic in these textiles. Šoltys and colleagues [20] sequenced the ITS, 16S and 28S rDNA regions to study the biodeteriogenic microbiome of a XVIII Century wax seal colored with minium. The authors identified the presence of a complex microbiota dominated by fungi capable of conducting alterations to lipids, leads, and contributing to the formation of lead soaps and secondary biogenic minerals. Piñar and colleagues [21,22] conducted two studies using a whole genome amplification (WGA) approach in various oil paintings and Leonardo da Vinci’s drawings. The authors were able to establish a fast and simple molecular WGA protocol for application in the area, while also highlighting microbiome variances in different composition and conservation status of distinct paintings [21]. For the Leonardo da Vinci’s drawings [22], they were able to observe the impacts of geographical region on the microbiome and obtain a microbiological “bio-archive”, and consequently a reference dataset for future monitoring efforts. Planý and colleagues [23] conducted a metabarcoding study of the 16S and 28S rDNA regions to understand iron nails corrosion from a whale skeleton displayed at the Natural History Museum in London, United Kingdom. Through this method, they were able to characterize the fungal and bacterial communities associated with the corrosion of the iron nails, while also proposing a fungal-mediated biodeterioration mechanism. Brimblecombe and colleagues [24] sequenced the ITS region to study the fungal surface contamination in the Klosterneuburg Monastic Library. The authors also compared these results with cultivated fungi retrieved from library dust at this site. Delegou and colleagues [25] applied 16S rDNA sequencing to characterize the microbiota of building materials of the holy Aedicule sepulcher in Jerusalem. They were also able to characterize and discuss the biodeteriorative impact of the major species found. Pavlović and colleagues [26] analyzed various genes (ITS and 16S rDNA region; and the nitrite reductase (nirK), sulfite reductase (dsr), the sox enzyme system for sulfur oxidation (soxB) and the ammonia monooxygenase (amoA) genes) to study a salt-contaminated twelfth century granite-built chapel. The authors contributed to the knowledge of the role of microorganisms in the presence of salt and damp stains, by founding a well-defined relationship between the microbiome and soluble salts, and consequently the biodeterioration phenomena affecting these chapels. Through the analysis of the ITS, 16S and 28S rDNA regions, Pavlović and colleagues [27] studied the microbiome of wax drippings from a candle on manuscripts. They found that wax did not make the paper more biodegradable, but provided niches for the accumulation of dust and eroded material, which in turn could act as nutritional hotspots for microbial proliferation. Rabbachin and colleagues [28] performed WGA sequencing in petroglyph sites of the Negev desert of Israel. The authors corroborated the connections of the microbial communities between rock varnish and stone inhabitants, and the microbiome contribution to the deterioration visualized. Timoncini and colleagues [29] conducted 16S rDNA sequencing to understand the role of microbial communities on bronze and marble statues patinas. They verified that differential bacterial communities occurred between marble and bronze statues and among different marble patinas. Additionally, the authors noted high microbial diversities in marble surfaces, low microorganism diversity in bronze statues, being able to expand the current knowledge regarding bacteria communities and various patinas. Beccaccioli and colleagues [30] targeted the ITS and 16S rDNA regions to understand the microbial communities contributing to the deterioration of waterlogged wooden fragments from pirogues. The authors also developed and tested a custom pipeline for their bioinformatics analysis. The results obtained allowed to identify high levels of many bacteria and ligninolytic fungi able to contribute to wood erosion of these artifacts. Li and colleagues [31] used a WGA approach (Illumina + Nanopore) in biofilms thriving in Leshan and Feilaifeng stone cultural heritage sites. They were able to understand the biofilms’ taxonomic profiles, while also gathering a strong knowledge of the microbial groups and gene families contributing to elemental nitrogen and sulfur metabolism enhancing stone biodeterioration. In addition, the authors were also able to shed light on biofilm resistome (see below). Nir and colleagues [32] applied the Oxford Nanopore® MinION™ sequencer to gather knowledge on the genomic characteristics of cyanobacteria colonizing the Negev petroglyphs. The authors were able to generate a metagenome from Trichocoleus desertorum strain NBK24 to understand genes crucial for survival at these extreme environments and contributing to elemental exchanges leading to biodeterioration. Pavlović and colleagues [33] conducted a metabarcoding analysis of 16S and 28S rDNA regions to study the microbiome of books, folklore prints and archive documents. The authors noted that complex microbial communities were responsible to stain two types of paper analyzed. Rabbachin and colleagues [34] applied metagenomic sequencing (WGA) to study natural patinas in petroglyphs in the Austrian Alps. They found differences on stone microbiomes with and without visible biofilm/patinas and concluded that their removal could enhance and continue new cycles of colonization and biodeterioration. Through the metabarcoding analysis of the 16S rDNA region, Tichy and colleagues [35] were able to understand the relationships between the microbiome and pinkish patinas in salt-weathered buildings. The authors found that the microbial communities were influenced by salt concentration and chemical composition. Moreover, this study also highlighted the impact of presence/absence of K+ ions on the bacteria and archaea communities and consequently biodeterioration at these sites. Bastholm and colleagues [36] sequenced the calmodulin gene through metabarcoding approaches to identify xerophilic Aspergillus growth in Danish Museum repositories. Comparisons with Illumina amplicon sequencing and cultivation-dependent methods were also performed, in an effort to understand if Aspergillus section Restricti species had become a novel contaminant nationally distributed. Haedar and colleagues [37] conducted analysis of the 16S rDNA region to understand the bacterial communities on degraded Prehistoric rock paintings in Maros-Pangkep Global Geopark. They found multiple genera known to be involved in carbonate precipitation and rock weathering contributing to the problems noted at this site. Rabbachin and colleagues [38] examined the fungal diversity at petroglyph sites in the Negev Desert, Israel. They compared mycobiomes from stone, soil and air, discovering a high fungal biodiversity in the soil, a dominance of Cladosporium in the air and the presence of lichens and extremotolerant microcolonial fungi in the stone materials.
Overall and taking into account these works, it is clear that the Oxford Nanopore® sequencer is increasingly becoming a critical component of microbial cultural heritage biodeterioration and conservation studies [8,9,17,39]. Some trends in the application of this system can also be verified. The number of studies using a metabarcoding approach are predominant, with the most sequenced genes being the ITS, 16S and 28S rDNA regions. In parallel, the most studied supports are related to stone substrates (Figure 2). The most common keywords found in these works include: biodeterioration, metagenomics, nanopore sequencing technology, building materials, stone, microbial community, 16s RNA ribosomal gene, microbiome, bacteria, nanopore sequencing technology, MinION™, microbiota and conservation (Figure 3). Additionally, through the analysis of the most common words found in the abstracts of these manuscripts (Figure 4), some of the Oxford Nanopore® MinION™ additional tendencies can also be further understood. In fact, a great deal of focus of these works is given to microorganism abilities to grow in museum environments and drawings, the biodeterioration of stone substrates affected by salts and the resistance mechanisms of biodeteriogens.
As such, some consequently current gaps of the application of the technique in this area include the need: (1) to expand the number of studies in supports less studied or not yet explored; (2) to conduct further discussions and comparisons regarding bioinformatic pipelines and analysis; and (3) to proceed with the expansion of application of WGA tailored studies, in order to deepen the knowledge of metabolic and resistance characteristics of microorganisms. Moreover, other additional topics could also be further investigated, such as the case of biodeteriogens metatranscriptomics, microbial ecology, on-site real-time application and molecular monitoring; biotechnology focusing in preservation/restoration efforts; and the monitoring of treatment efficiency through time.
3. Future Directions for Oxford Nanopore® Applications in Biodeterioration Studies
Pavlović and colleagues [17] provided an initial review of the first five articles where the technique was applied in this area [18,19,20,21,22]. The authors also discussed that future works could focus on the study of cultural heritage microbiomes metabolic and degradative features, the further creation of biobanks, the impacts of climate change on microbes, bioinformatics difficulties and available pipelines, and the improvements being made to the device sequencing capacities.
However, during the last five years, the technique application in various differential contexts has also opened the doors for their putative usage on other interesting sub-areas of research. These include, for instance: biodeteriogens genome and transcriptome analysis, metatranscriptomics, biodeteriorative metabolism studies, inter and intra kingdom interactions analysis, resistome understanding, biocodicology, object history and context, in situ applications, bioprospecting and biotechnology (Figure 5).
3.1. Biodeteriogens Genomics and Transcriptomics
Genome-wide and transcriptomic analysis can provide valuable information regarding the genetic basis and microbial mechanisms involved in biodeterioration. Nonetheless, one of the current major challenges for advancements in this area is the lack of sequenced microbial genomes and the absence of transcriptomic experimental data. For example, for black fungi (one of the major problematic microorganisms in stone), only a very low number of genomes from isolates retrieved from stone monuments are currently available [40,41]. Additionally, the number of available transcriptomic studies in the area, is much lower.
In a recent study, Quach and colleagues [42] demonstrated the utility of genome sequencing to understand lifestyle adaptations and glass biodeterioration mechanisms of Curvularia eragrostidis with the Illumina technique. The authors were able to characterize the metabolic pathways involved in adaptations to the species glassy habitat and the genomic basis for organic acid, exopolysaccharide biosynthesis and enzymatic production (products able to conduct biodeterioration).
Paiva and colleagues [43] also used Illumina to sequence the genome of the microcolonial black fungus Saxispiralis lemnorum and conducted a deep genomic analysis of the family Aeminiaceae. The authors found, not only various peculiar features for extremotolerance, but also metabolic properties that can putatively contribute to biodeterioration, such as enzymatic characteristics and their abilities for nitrate assimilation and sulfate reduction.
Pei and colleagues [44] conducted studies on the genomic and transcriptomic characteristics of Naumannella cuiyingiana AFT2T contributing to the lead-containing pigment discoloration of a 1500 years tomb wall painting of China. The authors found that the Pb ions presence in culture media induced changes in gene expression related to metal ion transport and metabolism genes, and to TlpA family protein disulfide reductases. Enzymes with disulfide-bond reducing properties and genes encoding the divalent Pb ion uptake transporter were also highlighted as the key genes in the process of discoloration of this tomb wall.
Wang and colleagues [45] studied the biodegradation mechanism of Fusarium solani NK-NH1 on the hull wood of the Nanhai No. 1 shipwreck. The authors performed whole genome sequencing of this isolate and were able to characterize the key genes responsible for cellulose and lignin degradation.
Presently, the Oxford Nanopore® technique has been shown to be able to sequence complete bacterial and plasmid genomes without the need for short-read sequencing [46]. Complementarily, the method has also been shown to be able to obtain near-complete, telomere to telomere fungal genomes [47,48,49] and with the current advancements in flow cell technology, will surely allow for improved results in a near future. Nonetheless, neither biodeteriogenic microorganisms genomic or transcriptomic analyses have been conducted with this system in the area. Such approach could be an interesting and useful application for the technique in future studies, especially considering the long-read capacities of the device. Their application could potentially contribute (along with metatranscriptomics) to provide insights on one of the major questions of the area “What do organisms do on and with the object?” [8].
3.2. Metatranscriptomics
While a large number of studies in the area using metagenomics or metabarcoding are available, there is still a lack of metatranscriptomics works focusing on cultural heritage biodeterioration scenarios. As pointed by Sterflinger and Piñar [8], this approach could offer a huge advancement in the area since it would allow to understand the biodeteriogenic and metabolic impact of microorganisms in an object under certain conditions. Zhang and colleagues [50] have studied the active RNA-level community at the Beishiku Temple in China using the Illumina data. The authors found a congruence in the results obtained through metagenomics and metaproteomic approaches and confirmed the impact of cyanobacteria in newly formed biofilms on stone monuments at this site. However, the Oxford Nanopore® sequencer has yet to be implemented in the area for metatranscriptomics study, a topic that is highly worthy of investigations, since the sequencer has been showed to perform such types of analysis even in unusual conditions such as the ones found at the International Space Station (ISS) [51].
3.3. Biodeteriorative Metabolic Insights
While a considerable number of studies employing the sequencer have been conducted with a WGA approach (Table 1), mainly the works of Li and colleagues [32] and Nir and colleagues [33] dwelled deeper in the biodeteriorative metabolism potential from the metagenomic analysis data obtained. In the first work, this approach allowed to deeply understand metabolic pathways contributions to biogeochemical N and S cycling, known to deeply enhance stone biodeterioration [52]. The second work, focused on obtaining and characterizing the metagenome of a cyanobacteria. The results allowed to characterize genes involved in element exchange, namely ATPases and membrane transporters with potential role in weathering processes.
It is clear that even with other NGS sequencing techniques [53,54], such studies are critical for understanding not only the microbial agents responsible for biodeterioration, but also their mechanisms of action. For this reason, future studies with this sequencer in these topics can also offer a wide range of possibilities and help answer critical questions in the area (how biodeterioration occurs in each specific setting).
3.4. Multi-Kingdom Interactions and Microbial Ecology
Biodeterioration of cultural heritage materials can occasionally occur due to the impact of one or a restricted group of microorganisms, although the majority of these processes are a result from a complex microbial community that establishes and displays intra- and inter-species associations and interactions [55,56]. Indeed, multi-kingdom mutualistic, competitive and neutral relationships can shape the cultural heritage materials microbiome [55,56], and consequently the biodeterioration processes affecting them. Thus, a deeper understanding of ecological networks can also provide fundamental knowledge on biodeteriorative microbes, contributing to predict their assemblies and help in their control [56].
Liu and colleagues [55] found that multi-kingdom interactions governed the microbiome in the subterranean Chinese Dahuting Han Dynasty Tomb. The authors found that Actinobacteria and Pseudonocardiaceae, by producing volatile geosmin, could attract Colembola that contributed to their dispersion to the tomb interior from the surrounding environment (inter-kingdom mutualism). The results also highlighted that intra-kingdom competition also occurred, as Pseudonocardiaceae thrive due to the production of cellulases and potential antimicrobial substances.
Yu and colleagues [56] conducted a metabarcoding meta-analysis of ~1000 microbiomes from cultural heritage sites in contrasting environmental conditions. The authors found that, at a global scale, bacterial communities are mainly influenced by global climate, while fungal communities’ dominance is largely explained by local habitat conditions.
In one of our recent studies in a marble statue [57], we also found some peculiar results, since multiple negative correlations were found between Bacteria and Fungi at this monument. Considering that the majority of Bacteria detected were Cyanobacteria, more positive correlations were expected, since they contribute to heterotrophs development.
Considering the importance and need to understand intra- and inter-species ecological interactions to improve long-term conservation of cultural heritage materials, future studies with the Oxford Nanopore® sequencer (using metagenomics and metatranscriptomics simultaneously, for instance) could provide critical knowledge to understand both the microbiome and their metabolic traits contributing to these interactions.
3.5. Cultural Heritage Resistome
The cultural heritage resistome, i.e., the antimicrobial resistance genes (AMR or ARGs) of a community, is a topic gaining some recent focus on the area [31,58,59]. Studies on AMR genes on cultural heritage monuments and relics can be considered of great relevance, considering that they can allow to understand anthropogenic activity impacts, to monitor resistance to preservation/conservation efforts, to understand contamination source tracking and even to contribute to public health management [31,58,59].
Li and colleagues [31] used both the Illumina and the Oxford Nanopore® techniques to understand the microbiome and key genetic contexts from Leshan and Feilaifeng stone heritages. The authors applied the Oxford Nanopore® sequencer for the first time in this context, and found high abundances and diversity of antibacterial biocide and metal resistance genes to copper and quaternary ammonium compounds. Furthermore, they also verified that diverse antimicrobial resistance genes and mobile genetic elements can be transmitted through horizontal gene transfer, putatively difficulting treatment applications on these relics. A comparison between both methods is also provided by the authors, confirming the ability to: (1) obtain average longer read lengths (8510 bp vs 304,680 bp from Illumina); (2) improved contig results (9390 vs 542,843 bp from Illumina) with the Oxford Nanopore® sequencer; and (3) an overall higher quality average contig N50 value, than when solely applying the Illumina reads [31].
He and colleagues [58] studied the environmental resistome and mobilome from Feilaifeng stone heritage sites in Hangzhou, Zhejiang Province in China, with the Illumina technique. The authors found that abundant and diverse AMR genes confer resistance to drugs (antibiotics), biocides and metals in the substrates studied, and that anthropogenic activities likely impact stone resistomes across stone heritage areas. Moreover, the authors also found that stone resistome and mobilome can improve their adaptation and confer resistance to antimicrobials applied against biodeterioration.
Ding and colleagues [59] studied pathogenicity characteristics and resistome profiles of the microbiome on the Angkor sandstone monuments in Cambodia with the Illumina technique. The authors found a specific set of ARGs with cross-niches between the environment and warm blood fauna. Additionally, they also verified resistance mechanisms on these supports and discussed their implications for public health on tourism of cultural heritage sites.
Considering the importance and need to understand resistance mechanisms of treatments applied on the conservation of cultural heritage materials and the AMR treat to public health, more studies with this unique sequencer could provide a deeper knowledge for the safety of visitors, contributing towards the one-health perspective and to improve conservation efforts. Recently, the technique alone was proven to be a quick, easy and reliable method for their detection [60], and their application on the area will surely have a further deep impact of our understanding of the microbiome dynamics and resistance by monitoring treatment effectivity.
3.6. Cultural Heritage Objects History
Molecular studies have also been shown to provide important information regarding the history, context, manufacturing materials, storage conditions, geographical origin and tampering of art objects [21,22,61,62]. In addition, they can also provide important clues about interactions of the relics with human interactions [61]. As such, they can provide important information in fields such as biocodicology, archeology, criminology but also in human contamination from handling [21,22,62,63,64]. While an emerging topic, attempts to apply the Oxford Nanopore® sequencer for the identification of the microbiome and animal ancient DNA (aDNA) from a 15th-century parchment from the Graphic Collection (Kupferstichkabinett) of the Academy of Fine Arts, in Vienna [62], have also been conducted. On the other hand, their application can also enhance our understanding of the object context and to help shedding light on the biodeteriorative/bioprotective dualistic effect/nature of the microbiome [64]. Understandably, this sub-area can also open doors for multiple applications of the Oxford Nanopore® sequencer worthy of further research.
3.7. On-Site Application
Due to the sequencer portability, handling features, adaptability and its analysis speed, the platform allows for an on-site analysis. In fact, multiple reports of such applications are currently available and the technique has been applied for real-time sequencing in differential environmental contexts, such as Ebola surveillance in West Africa, water quality monitoring, environmental microbe analysis, coronavirus SARS-CoV-2 diagnosis and even aboard the ISS [65,66,67,68,69,70,71,72,73]. The technique has also demonstrated to be reliable in resource-limited settings [71], a condition that is also verified for some cultural heritage monuments worldwide. In addition, Oxford Nanopore® WGS analysis can also contribute to developing countries study of their historical properties’ microbial threats. However, the on-site capabilities have yet to be explored in this research area.
Recently, Tamames and colleagues [73] developed and tested in volcanic rocks in La Palma, Canary Islands and marine waters, an in-situ system for Oxford Nanopore® metagenomic analysis in less than one day. Understandably, with such progress, the application of the technique for on-site analysis can offer a wide range of possibilities in the cultural heritage biodeterioration area, such as the monitoring of microbial outbreaks and in the implementation of quick control measures [74,75].
Complementarily, while culture-dependent methodologies face limitations to provide a holistic perspective of the microbiome causing biodeterioration of an artifact, they can offer important information since they allow microbe isolation for differential analysis, novel microorganisms discovery and the improvement of biological databases [76,77]. The sequencer has also been shown to be effective for in situ bioprospecting applications and microbial characterization [78], which considering the often-uncharacterized components of heritage materials microbiome [77], could also be worthy of future investigations.
3.8. Biotechnology and Restoration Efforts
From a biotechnological perspective, green intervention methodologies such as bioconsolidation and biocleaning [77] could also benefit of their coupling with the sequencer. Bioconsolidation refers to the set of methodologies consisting in the application of bacteria to remediate building materials, by mimicry of bacterial precipitation of carbonates on carbonate rocks [80,81]. On the other hand, biocleaning, consists in the exploration of microbial metabolism properties that act on removal of sulphates, nitrates and organic matter, in order to clean or ameliorate affected stone surfaces [82]. In this topic, the Oxford Nanopore® sequencer could be used to explore and to further understand bioconsolidation and biocleaning mechanisms through genomic and transcriptomic analysis which in turn could open doors for genomic manipulation and enhancement of these properties. Additionally, contributions towards the possible application and improvement of alternative microbial derived strategies such as the application of antagonistic microorganisms or even bacteria able conduct to bioremoval, could also benefit from this system [83,84,85,86]. Lastly, application of the method to understand the effectiveness of biocide treatments can also be a promising outlook, as it has already been demonstrated to be useful with the Illumina system [87].
4. Challenges in the Oxford Nanopore® Application in Biodeterioration Studies
While the sequencer presents numerous advantages for studying microbial communities in cultural heritage materials, several limitations must also be acknowledged. Among them, it should be highlighted that the relative high costs for some researchers, the difficulties to attain proper reagent storage in low-resource or remote settings and the need for strong internet connections for analysis, can hamper new applications of the method [71].
On the other hand, the occurrence of sequencing errors and the complexity of bioinformatics pipelines required for data analysis can also present further challenges for researchers sing this technique. Currently, while not error-free, much of the long-read associated issues have largely been solved due to the development of multiple error correction software’s in the last years [88] and to the creation of new flow cells. In fact, the Oxford Nanopore® systems are one of the fastest developing NGS platforms today and their limitations are systematically being improved. For instance, the new flow cell R10.4.1 allows quality scores of Q20+, very high accuracy levels (often over 99%) and even allows Short Fragmented Mode (SFM) applications [62,89,90,91,92] which, in itself, can offer another additional range of possibilities.
Less consensus occurs, however, when dealing with the bioinformatic analysis of the data obtained. Current limitations and criticisms include: the need for user-friendly bioinformatics platforms; the large amount of data obtained that can lead to enormous IT resource demands [72]; and the availability of methods and comprehensive databases for these analyses [87]. Moreover, the system standard bioinformatic platforms such as Epi2me requires internet connection, although offline applications are currently also available [93]. Nonetheless, current testing of alternative bioinformatic pipelines and databases in and outside of the CHB area [18,30,33,94,95,96,97,98,99,100,101] is an ongoing effort, which will surely require additional analysis and standardization in the near future.
Additionally, while MinION™ offers portability and in-field analysis capabilities, sample preparation protocols may need optimization to ensure reproducibility and reliable results in non-laboratory settings. On the other hand, and to a lesser degree, difficulties to conduct routinary procedures in situ, such as low DNA recovery from environmental samples, also needs to be noted [73]. While protocols for improved DNA extraction (e.g., [102]) have been developed, they need to be further tested in cultural heritage biodeterioration scenarios.
Overall, addressing these limitations will understandably be crucial to effectively ensure the utility of nanopore sequencing [88] in cultural heritage biodeterioration studies and in advancing our understanding of microbial interactions and degradation processes affecting cultural heritage artifacts.
5. Conclusions
So far, the application of Oxford Nanopore® MinION™ sequencing technology in cultural heritage biodeterioration studies has demonstrated a high potential and versatility. The technology has been effectively been applied in a wide range of materials, allowing the study of the diverse microbial communities and some metabolic pathways involved in their biodeterioration. Current trends in the area, include a tendency towards gene amplification tailored studies and a great focus in stone related materials. Future perspectives for the application of the technique in this field are promising, since due to the sequencer characteristics, they can be applied: for in situ analysis and bioprospecting; to microorganisms deeper genomic, transcriptomic and environmental metatranscriptomics analysis to understand active metabolic processes; to understand inter- and intra-kingdom microbial interactions and to understand objects history. Additionally, exploration of cultural heritage materials resistome can provide critical information on antimicrobial resistance, leading to the development and implement of improved conservation strategies. Furthermore, the technology's use in biotechnology could allow to the enhancement of innovative conservation methods. In conclusion, the sequencer has become an invaluable tool in the field of cultural heritage conservation and the exploration of new applications in the area can contribute to the preservation of cultural heritage relics and sites for future generations.
Author Contributions
Conceptualization, J.T.; formal analysis, J.T. and F.S.; investigation, J.T. and F.S.; writing—original draft preparation, J.T.; writing—review and editing, F.S.; project administration, J.T. and F.S.; funding acquisition, J.T.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Santander/University of Coimbra in the framework of the SeedProjects@UC 2024 with the reference PT0054.A.01.C. This work was carried out at the R&D Unit Centre for Functional Ecology—Science for People & the Planet (CFE) and Associate Laboratory TERRA. The Centre for Functional Ecology—Science for People and the Planet (CFE) was supported by FCT - Fundação para a Ciência e Tecnologia, I.P. by project reference UIDB/04004/2020 and the DOI identifier 10.54499/UIDB/04004/2020 (https://doi.org/10.54499/UIDB/04004/2020). The Associate Laboratory TERRA was supported by FCT - Fundação para a Ciência e Tecnologia, I.P. by project reference LA/P/0092/2020 and the DOI identifier 10.54499/LA/P/0092/2020 (https://doi.org/10.54499/LA/P/0092/2020). The authors also thank the funding of PRR—Recovery and Resilience Plan and by the NextGeneration EU European Funds.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hueck, H. J. The biodeterioration of materials as part of hylobiology. Mater Org, 1965, 1, 1. 5– 34.
- Hueck, H.J. , The biodeterioration of materials – an appraisal. In Biodeterioration of Materials. Walters, A.H., Elphick, J.S., Eds, Elsevier, London, UK, 1968, 6–12.
- Urzì, C. , Krumbein. W.E., Microbiological impacts on the cultural heritage. In Durability and change: the science, responsability and cost of sustaining cultural heritage. Krumbein, W.E., Brimblecombe, P., Cosgrove D.E., Stainforth, S., Eds, John Wiley & Sons Ltd., London, UK, 1994, 107–135.
- Sterflinger, K.; Piñar, G. Microbial Deterioration of Cultural Heritage and Works of Art--Tilting at Windmills? Appl Microbiol Biotechnol. 2013, 97, 9637–9646. [Google Scholar] [CrossRef] [PubMed]
- Favero-Longo, S.E.; Viles, H.A. A Review of the Nature, Role and Control of Lithobionts on Stone Cultural Heritage: Weighing-up and Managing Biodeterioration and Bioprotection. World J. Microbiol. Biotechnol. 2020, 36, 100. [Google Scholar] [CrossRef]
- Liu, X.; Qian, Y.; Wu, F.; Wang, Y.; Wang, W.; Gu, J.-D. Biofilms on Stone Monuments: Biodeterioration or Bioprotection? Trends Microbiol. 2022, 30, 816–819. [Google Scholar] [CrossRef]
- Beata, G. The Use of -Omics Tools for Assessing Biodeterioration of Cultural Heritage: A Review. J Cult Herit. 2020, 45, 351–361. [Google Scholar] [CrossRef]
- Sterflinger, K.; Piñar, G. Molecular-Based Techniques for the Study of Microbial Communities in Artworks. In Microorganisms in the Deterioration and Preservation of Cultural Heritage; Joseph, E., Ed.; Springer International Publishing: Cham. 2021, 59–77 ISBN 978-3-030-69411-1.
- Piñar, G.; Sterflinger, K. Natural Sciences at the Service of Art and Cultural Heritage: An Interdisciplinary Area in Development and Important Challenges. Microb Biotechnol. 2021, 14, 806–809. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.J.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim Biophys Acta. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat Rev Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Senbeta, B.; Worku, T.; Mohammed, A.A.; Senbeta, B.; Worku, T. Pacific bioscience sequence technology: Review. Int J Vet Sci Res. 2022, 8, 27–33. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Pavlovic, J.; Cavalieri, D.; Mastromei, G.; Pangallo, D.; Perito, B.; Marvasi, M. MinION Technology for Microbiome Sequencing Applications for the Conservation of Cultural Heritage. Microbiol Res. 2021, 247, 126727. [Google Scholar] [CrossRef]
- Grottoli, A.; Beccaccioli, M.; Zoppis, E.; Fratini, R.S.; Schifano, E.; Santarelli, M.L.; Uccelletti, D.; Reverberi, M. Nanopore Sequencing and Bioinformatics for Rapidly Identifying Cultural Heritage Spoilage Microorganisms. Front Mater. 2020, 7. [Google Scholar] [CrossRef]
- Kisová, Z.; Planý, M.; Pavlović, J.; Bučková, M.; Puškárová, A.; Kraková, L.; Kapustová, M.; Pangallo, D.; Šoltys, K. Biodeteriogens Characterization and Molecular Analyses of Diverse Funeral Accessories from XVII Century. Appl Sci. 2020, 10, 5451. [Google Scholar] [CrossRef]
- Šoltys, K.; Planý, M.; Biocca, P.; Vianello, V.; Bučková, M.; Puškárová, A.; Sclocchi, M.C.; Colaizzi, P.; Bicchieri, M.; Pangallo, D.; et al. Lead Soaps Formation and Biodiversity in a XVIII Century Wax Seal Coloured with Minium. Environ Microbiol. 2020, 22, 1517–1534. [Google Scholar] [CrossRef]
- Pinar, G.; Poyntner, C.; Lopandic, K.; Tafer, H.; Sterflinger, K. Rapid Diagnosis of Biological Colonization in Cultural Artefacts Using the MinION Nanopore Sequencing Technology. Int Biodeterior Biodegradation. 2020, 148. [Google Scholar] [CrossRef]
- Piñar, G.; Sclocchi, M.C.; Pinzari, F.; Colaizzi, P.; Graf, A.; Sebastiani, M.L.; Sterflinger, K. The Microbiome of Leonardo Da Vinci’s Drawings: A Bio-Archive of Their History. Front Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Planý, M.; Pinzari, F.; Šoltys, K.; Kraková, L.; Cornish, L.; Pangallo, D.; Jungblut, A.D.; Little, B. Fungal-Induced Atmospheric Iron Corrosion in an Indoor Environment. Int Biodeterior Biodegradation. 2021, 159, 105204. [Google Scholar] [CrossRef]
- Brimblecombe, P.; Sterflinger, K.; Derksen, K.; Haltrich, M.; Querner, P. Thermohygrometric Climate, Insects and Fungi in the Klosterneuburg Monastic Library. Heritage. 2022, 5, 4228–4244. [Google Scholar] [CrossRef]
- Delegou, E.; Karapiperis, C.; Hilioti, Z.; Chasapi, A.; Valasiadis, D.; Alexandridou, A.; Rihani, V.; Kroustalaki, M.; Bris, T.; Ouzounis, C.; et al. Metagenomics of the built cultural heritage: microbiota characterization of the building materials of the holy aedicule of the holy sepulchre in Jerusalem. 2022, CIENTIFIC CULTURE, 8(2), 59-83. [CrossRef]
- Pavlović, J.; Bosch-Roig, P.; Rusková, M.; Planý, M.; Pangallo, D.; Sanmartín, P. Long-Amplicon MinION-Based Sequencing Study in a Salt-Contaminated Twelfth Century Granite-Built Chapel. Appl Microbiol Biotechnol. 2022, 106, 4297–4314. [Google Scholar] [CrossRef]
- Pavlović, J.; Sclocchi, M.C.; Planý, M.; Ruggiero, D.; Puškárová, A.; Bučková, M.; Šoltys, K.; Colaizzi, P.; Riccardi, M.L.; Pangallo, D.; et al. The Microbiome of Candle Beeswax Drops on Ancient Manuscripts. Int Biodeterior Biodegradation. 2022, 174, 105482. [Google Scholar] [CrossRef]
- Rabbachin, L.; Piñar, G.; Nir, I.; Kushmaro, A.; Pavan, M.J.; Eitenberger, E.; Waldherr, M.; Graf, A.; Sterflinger, K. A Multi-Analytical Approach to Infer Mineral–Microbial Interactions Applied to Petroglyph Sites in the Negev Desert of Israel. Appl Sci. 2022, 12, 6936. [Google Scholar] [CrossRef]
- Timoncini, A.; Costantini, F.; Bernardi, E.; Martini, C.; Mugnai, F.; Mancuso, F.; Sassoni, E.; Ospitali, F.; Chiavari, C. Insight on Bacteria Communities in Outdoor Bronze and Marble Artefacts in a Changing Environment. Sci Total Environ. 2022, 850, 157804. [Google Scholar] [CrossRef]
- Beccaccioli, M.; Moricca, C.; Faino, L.; Reale, R.; Mineo, M.; Reverberi, M. The Neolithic Site “La Marmotta”: DNA Metabarcoding to Identify the Microbial Deterioration of Waterlogged Archeological Wood. Front Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Li, Q.; Wu, C.; He, J.; Zhang, B. Unraveling the Microbiotas and Key Genetic Contexts Identified on Stone Heritage Using Illumina and Nanopore Sequencing Platforms. Int Biodeterior Biodegradation. 2023, 185. [Google Scholar] [CrossRef]
- Nir, I.; Barak, H.; Rabbachin, L.; Arielle, K.; Pavan, M.; Winter, E.; Pinar, G.; Sterflinger, K.; Ariel, K. Trichocoleus Desertorum Isolated from Negev Desert Petroglyphs: Characterization, Adaptation and Bioerosion Potential. The Sci Total Environ. 2023, 904, 166739. [Google Scholar] [CrossRef]
- Pavlović, J.; Puškárová, A.; Planý, M.; Farkas, Z.; Rusková, M.; Kvalová, K.; Kraková, L.; Bučková, M.; Pangallo, D. Colored Stains: Microbial Survey of Cellulose-Based and Lignin Rich Papers. Int J Biol Macromol. 2023, 241, 124456. [Google Scholar] [CrossRef]
- Rabbachin, L.; Pinar, G.; Nir, I.; Kushmaro, A.; Eitenberger, E.; Waldherr, M.; Graf, A.; Sterflinger, K. Natural Biopatina on Historical Petroglyphs in the Austrian Alps: To Clean or Not to Clean? Int Biodeterior Biodegradation. 2023, 183, 105632. [Google Scholar] [CrossRef]
- Tichy, J.; Waldherr, M.; Ortbauer, M.; Graf, A.; Sipek, B.; Jembrih-Simbuerger, D.; Sterflinger, K.; Piñar, G. Pretty in Pink? Complementary Strategies for Analysing Pink Biofilms on Historical Buildings. Sci Total Environ. 2023, 904, 166737. [Google Scholar] [CrossRef]
- Bastholm, C.J.; Andersen, B.; Frisvad, J.C.; Oestergaard, S.K.; Nielsen, J.L.; Madsen, A.M.; Richter, J. A Novel Contaminant in Museums? A Cross-Sectional Study on Xerophilic Aspergillus Growth in Climate-Controlled Repositories. Sci Total Environ. 2024, 173880. [Google Scholar] [CrossRef]
- Haedar, N.; Iqram, M.; Priosambodo, D.; Lebe, R. Bacterial Communities on Degraded Prehistoric Rock Paintings in Maros-Pangkep Global Geopark. Philippine Journal of Science, 2024, 153(1): 391-402.
- Rabbachin, L.; Nir, I.; Waldherr, M.; Vassallo, Y.; Piñar, G.; Graf, A.; Kushmaro, A.; Sterflinger, K. Diversity of Fungi Associated with Petroglyph Sites in the Negev Desert, Israel, and Their Potential Role in Bioweathering. Front Fungal Biol 2024, 5. [Google Scholar] [CrossRef]
- Marvasi, M.; Pangallo, D.; Cavalieri, D.; Poyatos-Jiménez, F. Editorial: Multi-Omics Revolution in Microbial Cultural Heritage Conservation. Front Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- De Leo, F.; Marchetta, A.; Urzì, C. Black Fungi on Stone-Built Heritage: Current Knowledge and Future Outlook. Appl Sci. 2022, 12, 3969. [Google Scholar] [CrossRef]
- Trovão, J.; Tiago, I.; Soares, F.; Paiva, D.S.; Mesquita, N.; Coelho, C.; Catarino, L.; Gil, F.; Portugal, A. High-Quality Draft Genome Sequence of the Microcolonial Black Fungus Aeminium Ludgeri DSM 106916. Microbiol Resour Announc. 2019, 8, e00202–19. [Google Scholar] [CrossRef]
- Quach, N.T.; Ngo, C.C.; Nguyen, T.H.; Nguyen, P.L.; Vu, T.H.N.; Phan, T.H.T.; Nguyen, Q.H.; Le, T.T.M.; Chu, H.H.; Phi, Q.-T. Genome-Wide Comparison Deciphers Lifestyle Adaptation and Glass Biodeterioration Property of Curvularia Eragrostidis C52. Sci Rep. 2022, 12, 11411. [Google Scholar] [CrossRef]
- Paiva, D.S.; Fernandes, L.; Portugal, A.; Trovão, J. First Genome Sequence of the Microcolonial Black Fungus Saxispiralis Lemnorum MUM 23.14: Insights into the Unique Genomic Traits of the Aeminiaceae Family. Microorganisms. 2024, 12, 104. [Google Scholar] [CrossRef]
- Pei, S.; Wu, F.; Chen, Y.; Ma, W.; He, D.; Zhang, Q.; Gu, J.-D.; Wang, W.; Tian, T.; Feng, H. Mechanisms of Lead-Containing Pigment Discoloration Caused by Naumannella Cuiyingiana AFT2T Isolated from 1500 Years Tomb Wall Painting of China. Int Biodeterior Biodegradation. 2023, 185, 105689. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Y.; Li, N.; Wang, C.; Ma, K.; Huang, X.; Du, J.; Guo, H.; Pan, J. Study on Biodegradation Mechanism of Fusarium Solani NK-NH1 on the Hull Wood of the Nanhai No. 1 Shipwreck. Front Microbiol. 2024, 15, 1382653. [Google Scholar] [CrossRef]
- Zhao, W.; Zeng, W.; Pang, B.; Luo, M.; Peng, Y.; Xu, J.; Kan, B.; Li, Z.; Lu, X. Oxford Nanopore Long-Read Sequencing Enables the Generation of Complete Bacterial and Plasmid Genomes without Short-Read Sequencing. Front Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Salazar, A.N.; Gorter de Vries, A.R.; van den Broek, M.; Wijsman, M.; de la Torre Cortés, P.; Brickwedde, A.; Brouwers, N.; Daran, J.-M.G.; Abeel, T. Nanopore Sequencing Enables Near-Complete de Novo Assembly of Saccharomyces Cerevisiae Reference Strain CEN.PK113-7D. FEMS Yeast Res, 2017, 17, fox074. [CrossRef]
- McGinnis, J.L.; Giguere, D.J. High-Quality Genome Assembly of a Pestalotiopsis Fungus Using DIY-Friendly Methods 2022. [version 1; peer review: 3 approved with reservations]. F1000Research, 2022, 11:442 . [CrossRef]
- Witte, T.E.; Hicks, C.; Shoukouhi, P.; Dadej, K.; Findlay, W.; Liu, M.; Overy, D.P. Chromosome-Level Draft Genome Sequences of Three Isolates of the Toxigenic Fungus Claviceps Purpurea Showing Structural Rearrangements. Microbiol Resour Announc. 2023, 12, e00234–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, F.; Gu, J.-D.; He, K.; Fang, Z.; Liu, X.; He, D.; Ding, X.; Li, J.; Han, Z.; et al. Dominance by Cyanobacteria in the Newly Formed Biofilms on Stone Monuments under a Protective Shade at the Beishiku Temple in China. Env Res. 2024, 251, 118576. [Google Scholar] [CrossRef]
- Haveman, N.J.; Khodadad, C.L.M.; Dixit, A.R.; Louyakis, A.S.; Massa, G.D.; Venkateswaran, K.; Foster, J.S. Evaluating the Lettuce Metatranscriptome with MinION Sequencing for Future Spaceflight Food Production Applications. npj Microgravity. 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Liu, X.; Koestler, R.J.; Warscheid, T.; Katayama, Y.; Gu, J.-D. Microbial Deterioration and Sustainable Conservation of Stone Monuments and Buildings. Nat Sustain. 2020, 3, 991–1004. [Google Scholar] [CrossRef]
- Wu, F.; Ding, X.; Zhang, Y.; Gu, J.-D.; Liu, X.; Guo, Q.; Li, J.; Feng, H. Metagenomic and Metaproteomic Insights into the Microbiome and the Key Geobiochemical Potentials on the Sandstone of Rock-Hewn Beishiku Temple in Northwest China. Sci Total Environ. 2023, 893, 164616. [Google Scholar] [CrossRef]
- Qian, Z.; Li, Y.; Pratush, A.; Kan, J.; Gu, J.-D.; Peng, T.; Huang, T.; Hu, Z. A Comparative Analysis of the Microbial Communities and Functional Genes of the Nitrogen Cycling in Mangroves of China, Indian and Malaysia. Int Biodeterior Biodegradation. 2024, 190, 105767. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, X.; Jin, T.; Li, Y.; Wu, B.; Yu, D.; Yu, Z.; Su, B.; Chen, R.; Feng, Y.; et al. Multikingdom Interactions Govern the Microbiome in Subterranean Cultural Heritage Sites. PNAS. 2022, 119, e2121141119. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, J.; Chen, R.; Coleine, C.; Liu, W.; Delgado-Baquerizo, M.; Feng, Y. Unearthing the Global Patterns of Cultural Heritage Microbiome for Conservation. Int Biodeterior Biodegradation. 2024, 190, 105784. [Google Scholar] [CrossRef]
- Trovão, J.; Portugal, A. The Impact of Stone Position and Location on the Microbiome of a Marble Statue. The Microbe. 2024, 2, 100040. [Google Scholar] [CrossRef]
- He, J.; Zhang, N.; Shen, X.; Muhammad, A.; Shao, Y. Deciphering Environmental Resistome and Mobilome Risks on the Stone Monument: A Reservoir of Antimicrobial Resistance Genes. Sci Total Environ. 2022, 838, 156443. [Google Scholar] [CrossRef]
- Ding, X.; Lan, W.; Li, J.; Deng, M.; Li, Y.; Katayama, Y.; Gu, J.-D. Metagenomic Insight into the Pathogenic-Related Characteristics and Resistome Profiles within Microbiome Residing on the Angkor Sandstone Monuments in Cambodia. Sci Total Environ. 2024, 918, 170402. [Google Scholar] [CrossRef]
- Solcova, M.; Demnerova, K.; Purkrtova, S. Application of Nanopore Sequencing (MinION) for the Analysis of Bacteriome and Resistome of Bean Sprouts. Microorganisms. 2021, 9, 937. [Google Scholar] [CrossRef]
- Piñar, G.; Poyntner, C.; Tafer, H.; Sterflinger, K. A Time Travel Story: Metagenomic Analyses Decipher the Unknown Geographical Shift and the Storage History of Possibly Smuggled Antique Marble Statues. Ann Microbio,l 2019, 69, 1001–1021. [Google Scholar] [CrossRef]
- Vassallo, Y.; Waldherr, M.; Lehner, E.; Graf, A.; Cappa, F.; Hartl, A.; Schober, R.; Beccaccioli, M.; Sterflinger, K.; Piñar, G.; et al. Oxford Nanopore Technologies for Biocodicology: A Case Study on a 15th-Century Parchment - Vassallo Y., Waldherr M., Lehner E., Graf A., Cappa F., Hartl A., Schober R., Beccaccioli M., Sterflinger K., Piñar G., Reverberi M. In; DTC Lazio: Tecnologie e patrimonio culturale: nuove competenze e professioni. 2023. https://iris.uniroma1.it/handle/11573/1695400?mode=complete.
- Simon, L.M.; Flocco, C.; Burkart, F.; Methner, A.; Henke, D.; Rauer, L.; Müller, C.L.; Vogel, J.; Quaisser, C.; Overmann, J.; et al. Microbial Fingerprints Reveal Interaction between Museum Objects, Curators, and Visitors. iScience. 2023, 26, 107578. [Google Scholar] [CrossRef]
- Cao, Y.; Bowker, M.A.; Delgado-Baquerizo, M.; Xiao, B. Biocrusts Protect the Great Wall of China from Erosion. Sci Adv. 2023, 9, eadk5892. [Google Scholar] [CrossRef]
- Castro-Wallace, S.L.; Chiu, C.Y.; John, K.K.; Stahl, S.E.; Rubins, K.H.; McIntyre, A.B.R.; Dworkin, J.P.; Lupisella, M.L.; Smith, D.J.; Botkin, D.J.; et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci Rep. 2017, 7, 18022. [Google Scholar] [CrossRef]
- Goordial, J.; Altshuler, I.; Hindson, K.; Chan-Yam, K.; Marcolefas, E.; Whyte, L.G. In Situ Field Sequencing and Life Detection in Remote (79°26′N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Front Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Isolation and Characterization of SARS-CoV-2 from the First US COVID-19 Patient. bioRxiv, 2020, 2020.03.02.972935.
- Latorre-Pérez, A.; Pascual, J.; Porcar, M.; Vilanova, C. A Lab in the Field: Applications of Real-Time, in Situ Metagenomic Sequencing. Biology Methods and Protocols. 2020, 5, bpaa016. [Google Scholar] [CrossRef]
- Moore, S.C.; Penrice-Randal, R.; Alruwaili, M.; Dong, X.; Pullan, S.T.; Carter, D.P.; Bewley, K.; Zhao, Q.; Sun, Y.; Hartley, C.; et al. Amplicon Based MinION Sequencing of SARS-CoV-2 and Metagenomic Characterisation of Nasopharyngeal Swabs from Patients with COVID-19 bioRxiv, 2020, 2020.03.05.20032011.
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-Time, Portable Genome Sequencing for Ebola Surveillance. Nature. 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Wasswa, F.B.; Kassaza, K.; Nielsen, K.; Bazira, J. MinION Whole-Genome Sequencing in Resource-Limited Settings: Challenges and Opportunities. Curr Clin Micro Rpt. 2022, 9, 52–59. [Google Scholar] [CrossRef]
- Werner, D.; Acharya, K.; Blackburn, A.; Zan, R.; Plaimart, J.; Allen, B.; Mgana, S.M.; Sabai, S.M.; Halla, F.F.; Massawa, S.M.; et al. MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research. Water. 2022, 14, 2491. [Google Scholar] [CrossRef]
- Tamames, J.; Jiménez-Lalana, D.; Redondo, Á.; Martínez-García, S.; De Los Rios, A. In Situ Metagenomics: A Platform for Rapid Sequencing and Analysis of Metagenomes in Less than One Day. Mol Ecol Resour. 2024, 24, e13909. [Google Scholar] [CrossRef]
- Bastholm, C.J.; Madsen, A.M.; Andersen, B.; Frisvad, J.C.; Richter, J. The Mysterious Mould Outbreak - A Comprehensive Fungal Colonisation in a Climate-Controlled Museum Repository Challenges the Environmental Guidelines for Heritage Collections. J Cult Herit. 2022, 55, 78–87. [Google Scholar] [CrossRef]
- Martin-Pozas, T.; Nováková, A.; Jurado, V.; Cuezva, S.; Fernandez-Cortes, A.; Saiz-Jimenez, C.; Sanchez-Moral, S. A Second Fungal Outbreak in Castañar Cave, Spain, Discloses the Fragility of Subsurface Ecosystems. Microb Ecol. 2024, 87, 53. [Google Scholar] [CrossRef]
- Trovão, J.; Portugal, A. Current Knowledge on the Fungal Degradation Abilities Profiled through Biodeteriorative Plate Essays. Appl Sci. 2021, 11, 4196. [Google Scholar] [CrossRef]
- Pyzik, A.; Ciuchcinski, K.; Dziurzynski, M.; Dziewit, L. The Bad and the Good—Microorganisms in Cultural Heritage Environments—An Update on Biodeterioration and Biotreatment Approaches. Materials. 2021, 14, 177. [Google Scholar] [CrossRef]
- Latorre-Pérez, A.; Gimeno-Valero, H.; Tanner, K.; Pascual, J.; Vilanova, C.; Porcar, M. A Round Trip to the Desert: In Situ Nanopore Sequencing Informs Targeted Bioprospecting. Front Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Andreolli, M.; Lampis, S.; Bernardi, P.; Calò, S.; Vallini, G. Bacteria from Black Crusts on Stone Monuments Can Precipitate CaCO3 Allowing the Development of a New Bio-Consolidation Protocol for Ornamental Stone. Int Biodeterior Biodegradation. 2020, 153, 105031. [Google Scholar] [CrossRef]
- Dhami, N.K.; Reddy, M.S.; Mukherjee, A. Application of Calcifying Bacteria for Remediation of Stones and Cultural Heritages. Front Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Reddy, M.S. Biomineralization of Calcium Carbonates and Their Engineered Applications: A Review. Front Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- Cappitelli, F. Biocleaning of Cultural Heritage Surfaces. Open Conf Proc J. 2016, 7. [Google Scholar] [CrossRef]
- Ranalli, G.; Zanardini, E. Biocleaning on Cultural Heritage: New Frontiers of Microbial Biotechnologies. Journal of Applied Microbiology. 2021, 131, 583–603. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Roig, P.; Sanmartín, P. Bioremoval of Graffiti in the Context of Current Biocleaning Research. In Microorganisms in the Deterioration and Preservation of Cultural Heritage; Joseph, E., Ed.; Springer International Publishing: Cham. 2021, 175–197 ISBN 978-3-030-69411-1.
- Cattò, C.; Sanmartín, P.; Gulotta, D.; Troiano, F.; Cappitelli, F. Bioremoval of Graffiti Using Novel Commercial Strains of Bacteria. Sci Total Environ. 2021, 756, 144075. [Google Scholar] [CrossRef]
- Sanmartín, P.; Bosch-Roig, P.; Pangallo, D.; Kraková, L.; Serrano, M. Unraveling Disparate Roles of Organisms, from Plants to Bacteria, and Viruses on Built Cultural Heritage. Appl Microbiol Biotechnol, 2023, 107, 2027–2037.
- Villar-dePablo, M.; Ascaso, C.; Rodríguez-Pérez, E.; Urizal, M.; Wierzchos, J.; Pérez-Ortega, S.; de los Ríos, A. Innovative Approaches to Accurately Assess the Effectiveness of Biocide-Based Treatments to Fight Biodeterioration of Cultural Heritage Monuments. Sci. Total Environ. 2023, 897, 165318. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Rodríguez-Pérez, H.; Flores, C. Nanopore Sequencing and Its Application to the Study of Microbial Communities. CSBJ 2021, 19, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, L.J. Is Oxford Nanopore Sequencing Ready for Analyzing Complex Microbiomes? FEMS Microbiol. Ecol. 2021, 97, fiab001. [Google Scholar] [CrossRef]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput Struct Biotechnol J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Ma, S.; Cao, J.; Liao, H.; Huang, Q.; Chen, W. The Newest Oxford Nanopore R10.4.1 Full-Length 16S rRNA Sequencing Enables the Accurate Resolution of Species-Level Microbial Community Profiling. Appl Environ Microbiol. 2023, 89, e0060523. [Google Scholar] [CrossRef]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 Long-Read Sequencing Enables the Generation of near-Finished Bacterial Genomes from Pure Cultures and Metagenomes without Short-Read or Reference Polishing. Nat Methods. 2022, 19, 823–826. [Google Scholar] [CrossRef]
- Zorz, J.; Li, C.; Chakraborty, A.; Gittins, D.A.; Surcon, T.; Morrison, N.; Bennett, R.; MacDonald, A.; Hubert, C.R.J. SituSeq: An Offline Protocol for Rapid and Remote Nanopore 16S rRNA Amplicon Sequence Analysis. ISME Commun. 2023, 3, 1–11. [Google Scholar] [CrossRef]
- Chandrakumar, I.; Gauthier, N.P.G.; Nelson, C.; Bonsall, M.B.; Locher, K.; Charles, M.; MacDonald, C.; Krajden, M.; Manges, A.R.; Chorlton, S.D. BugSplit Enables Genome-Resolved Metagenomics through Highly Accurate Taxonomic Binning of Metagenomic Assemblies. Commun Biol 2022, 5, 1–10. [Google Scholar] [CrossRef]
- Curry, K.D.; Wang, Q.; Nute, M.G.; Tyshaieva, A.; Reeves, E.; Soriano, S.; Wu, Q.; Graeber, E.; Finzer, P.; Mendling, W.; et al. Emu: Species-Level Microbial Community Profiling of Full-Length 16S rRNA Oxford Nanopore Sequencing Data. Nat Methods 2022, 19, 845–853. [Google Scholar] [CrossRef]
- Fan, J.; Huang, S.; Chorlton, S.D. BugSeq: A Highly Accurate Cloud Platform for Long-Read Metagenomic Analyses. BMC Bioinformatics 2021, 22, 160. [Google Scholar] [CrossRef]
- Jung, A.; Chorlton, S.D. BugSeq 16S: NanoCLUST with Improved Consensus Sequence Classification bioRxiv. 2021. [CrossRef]
- Petrone, J.R.; Rios Glusberger, P.; George, C.D.; Milletich, P.L.; Ahrens, A.P.; Roesch, L.F.W.; Triplett, E.W. RESCUE: A Validated Nanopore Pipeline to Classify Bacteria through Long-Read, 16S-ITS-23S rRNA Sequencing. Front. Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Planý, M.; Sitarčík, J.; Pavlović, J.; Budiš, J.; Koreňová, J.; Kuchta, T.; Pangallo, D. Evaluation of Bacterial Consortia Associated with Dairy Fermentation by Ribosomal RNA (Rrn) Operon Metabarcoding Strategy Using MinION Device. Food Bioscience 2023, 51, 102308. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, H.; Ciuffreda, L.; Flores, C. NanoCLUST: A Species-Level Analysis of 16S rRNA Nanopore Sequencing Data. Bioinformatics 2021, 37, 1600–1601. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, H.; Ciuffreda, L.; Flores, C. NanoRTax, a Real-Time Pipeline for Taxonomic and Diversity Analysis of Nanopore 16S rRNA Amplicon Sequencing Data. CSBJ 2022, 20, 5350–5354. [Google Scholar] [CrossRef]
- Maghini, D.G.; Moss, E.L.; Vance, S.E.; Bhatt, A.S. Improved High-Molecular-Weight DNA Extraction, Nanopore Sequencing and Metagenomic Assembly from the Human Gut Microbiome. Nat Protoc. 2021, 16, 458–471. [Google Scholar] [CrossRef]
Figure 1.
Example of an Oxford Nanopore® MinION™ sequencer device.

Figure 2.
Alluvial plot displaying a summary of works were Oxford Nanopore® MinION™ sequencer was applied in the cultural heritage biodeterioration field.
Figure 2.
Alluvial plot displaying a summary of works were Oxford Nanopore® MinION™ sequencer was applied in the cultural heritage biodeterioration field.
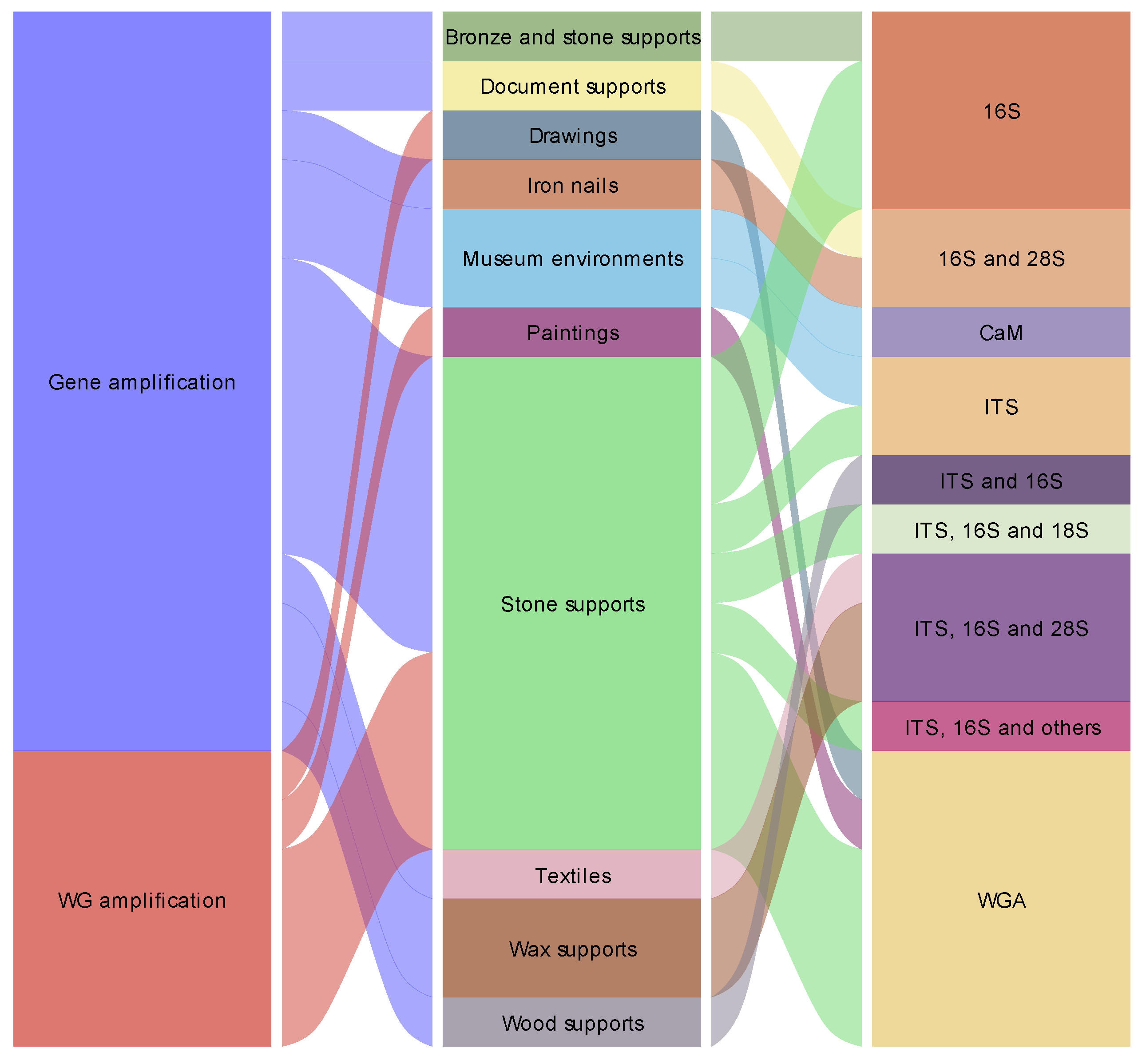
Figure 3.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main keywords (appearing at least two times) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
Figure 3.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main keywords (appearing at least two times) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.

Figure 4.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main words (appearing at least two times in the abstract section) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
Figure 4.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main words (appearing at least two times in the abstract section) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
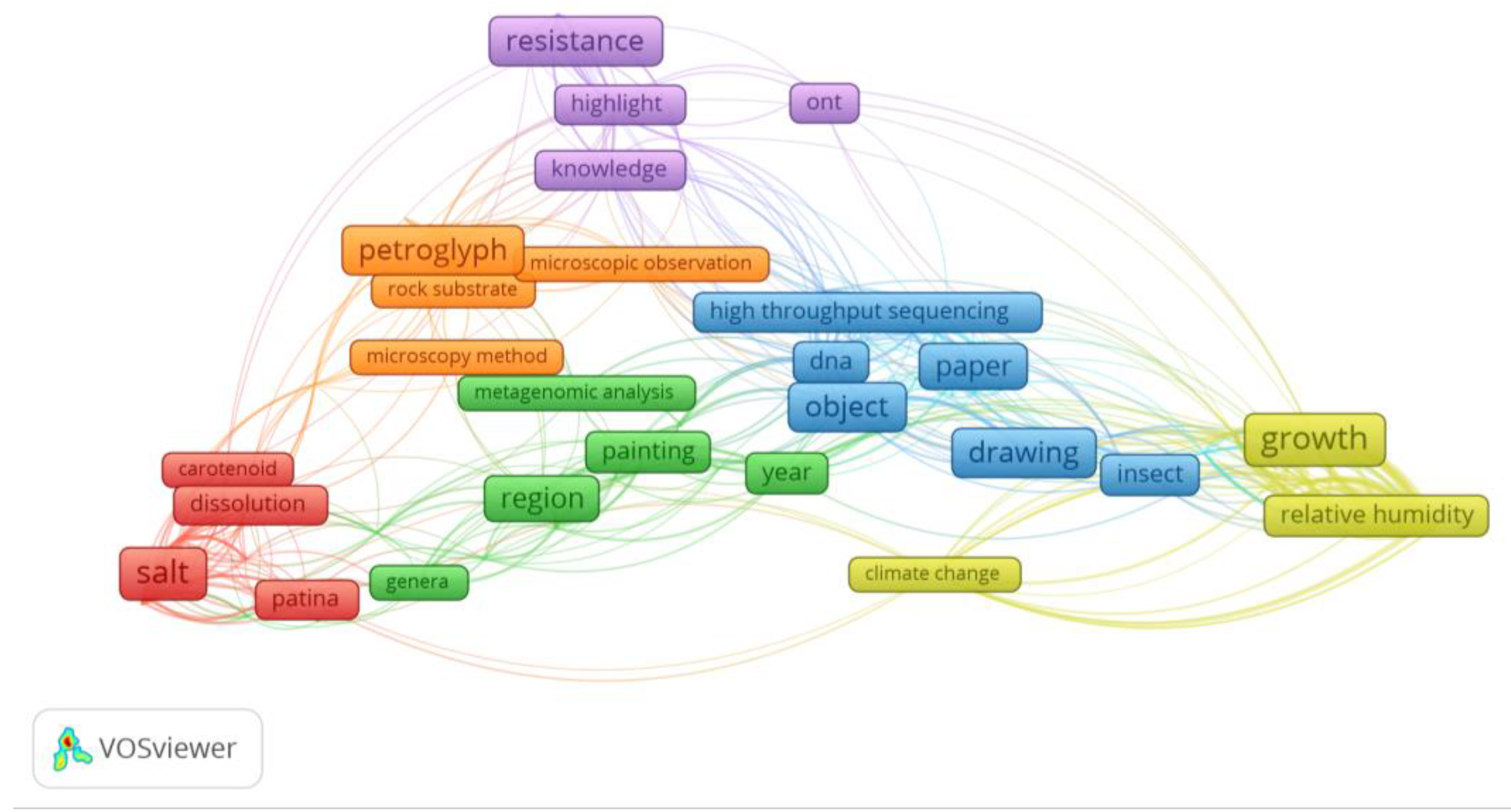
Figure 5.
Possible future directions for the Oxford Nanopore® application in biodeterioration studies.
Figure 5.
Possible future directions for the Oxford Nanopore® application in biodeterioration studies.
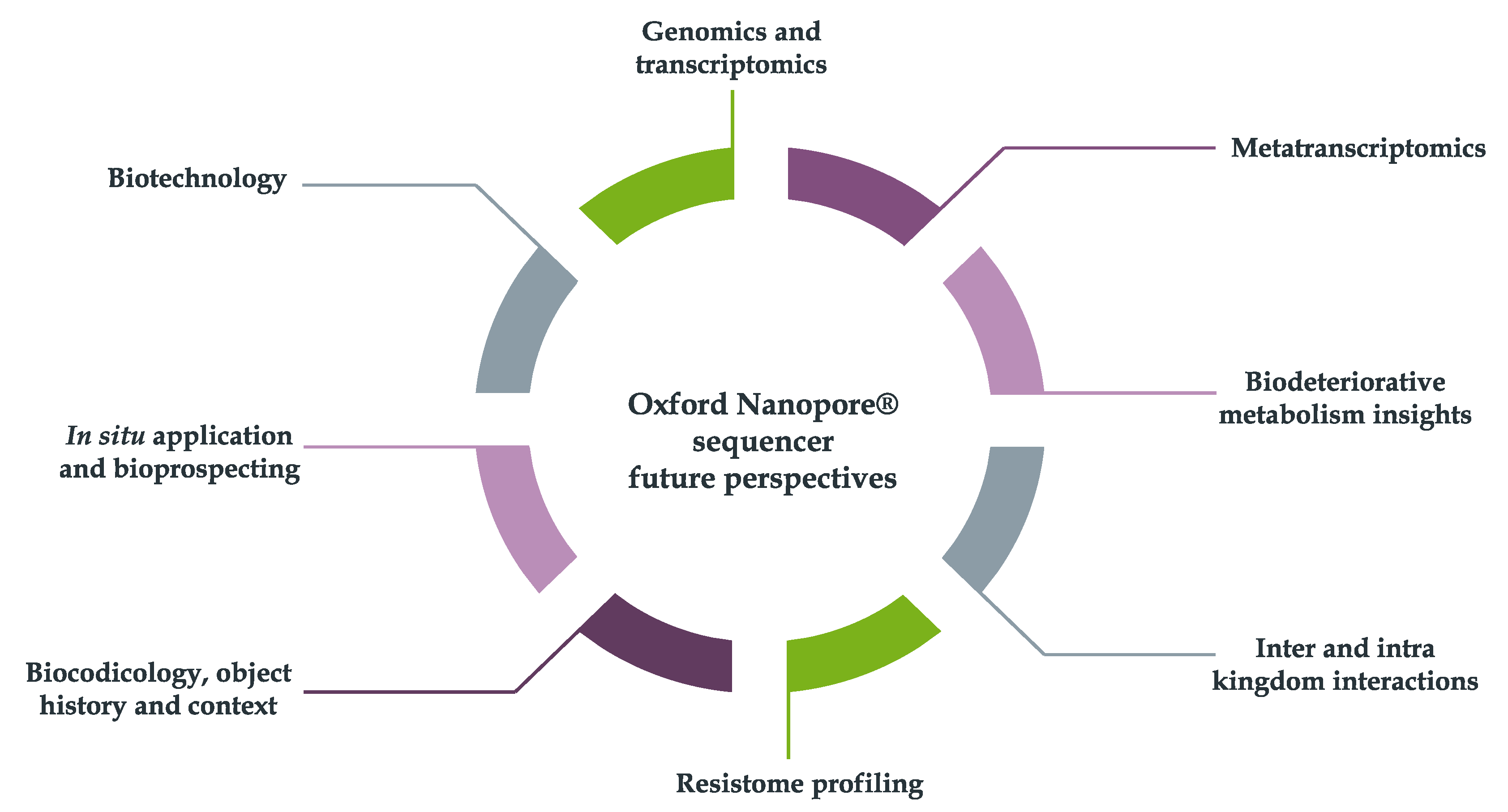

Table 1.
Cultural heritage biodeterioration studies conducted with the MinION™ sequencer.
| Approach | Material | Reference |
|---|---|---|
| Gene amplification | Stone monument | [18] |
| Gene amplification | Funeral accessories textiles | [19] |
| Gene amplification | Wax seal | [20] |
| Whole genome amplification | Oil painting | [21] |
| Whole genome amplification | Drawings | [22] |
| Gene amplification | Iron nails from whale skeleton | [23] |
| Gene amplification | Museum surfaces | [24] |
| Gene amplification | Stone monument | [25] |
| Gene amplification | Granite chapel | [26] |
| Gene amplification | Beeswax drops | [27] |
| Whole genome amplification | Petroglyph sites | [28] |
| Gene amplification | Bronze and marble statues | [29] |
| Gene amplification | Waterlogged wood | [30] |
| Whole genome amplification | Stone monument | [31] |
| Whole genome amplification | Petroglyph sites | [32] |
| Gene amplification | Documents | [33] |
| Whole genome amplification | Petroglyph sites | [34] |
| Gene amplification | Salt-weathered buildings | [35] |
| Gene amplification | Air and museum surfaces | [36] |
| Gene amplification | Rock Paintings | [37] |
| Gene amplification | Petroglyph sites | [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Submitted:
13 July 2024
Posted:
15 July 2024
Read the latest preprint version here
Alerts


This version is not peer-reviewed
Submitted:
13 July 2024
Posted:
15 July 2024
Read the latest preprint version here
Alerts
Abstract
The present work provides an updated overview of the application of the Oxford Nanopore® MinION™ sequencer in cultural heritage biodeterioration studies, while also providing a holistic discussion of possible future perspectives for their utilization in this research field. Due to the peculiar characteristics of this device, the last few years have seen the steady rise on the application of this system in a variety of cultural heritage materials, having been useful to understand microbial biodeteriogens diversity and some of their metabolic and biodeteriogenic features. Considering the immense potential for application of this system, this manuscript discusses further possibilities of the technique aiming to help understand critical questions on the cultural heritage biodeterioration area. Its application in various differential contexts, has opened the doors for their putative usage on other interesting sub-areas of research worthy of future investigations, including: biodeteriogens genome and transcriptome analysis, metatranscriptomics, biodeteriorative metabolism studies, inter and intra kingdom interactions analysis, resistome profiling, object history and context studies, in situ applications, bioprospecting and biotechnology.
Keywords:
Subject: Biology and Life Sciences - Life Sciences
1. Introduction
Humankind cultural heritage objects, relics and sites can be undesirably altered and seriously damaged from the growth and metabolic activities of living organisms [1,2,3]. These biodeterioration processes can occur at both indoor (e.g., museums) and outdoor environments (e.g., monuments), posing a serious risk for worldwide historical sites, properties and objects. Materials such as paper, ceramics, textiles, glass and stone, or objects including parchments, books, paintings, frescoes, vitrails, photographs, sculptures and funerary accessories, can be colonized and deteriorated through microorganisms’ aesthetic, mechanical, acid and enzymatic vital actions and manipulations [4]. Biodeterioration can thus be a result of the impact of various organisms (bacteria, cyanobacteria, microalgae, archaea, fungi and lichens [4]), and requires that protective measures are constantly considered, developed and implemented. Depending on specific conditions and the substrate type, some of these microorganisms can also contribute to the protection of the materials, thus displaying a biodeteriorative/bioprotective dualistic effect/nature [5,6]. For these reasons, molecular techniques, such as DNA sequencing, have been largely applied to investigate, understand and monitor biological colonization on art objects and cultural heritage monuments for more than two decades [7,8,9]. Justifiably, the current focus resides in the application of Next-Generation-Sequencing (NGS) methodologies, since they have powerfully expanded the possibility to characterize microbial communities in a cheaper, quick and holistic manner [10,11,12,13,14,15,16]. Due to the peculiar characteristics of this device, the last few years have seen the steady rise of the popular Oxford Nanopore® MinION™ sequencer in this research area, which has been applied in a variety of cultural heritage materials [8,9,17]. MinION™ is a small (Figure 1), mobile, relatively inexpensive, long read DNA/RNA sequencer, with an enormous range of applications. The technology relies on nanopores (protein pores), acting as biosensors to detect negatively charged single-stranded DNA or RNA molecules driven through the nanopore along an ionic current, and decoding of electric variations with computational algorithms [16]. In this review article, we aim to provide a brief update summary of previous studies using the MinION™ sequencer, while simultaneously providing a holistic discussion of possible future directions, additional applications and associated impacts of their utilization in the field of cultural heritage materials biodeterioration (in order to stimulate further discussions).
2. Application of Oxford Nanopore® in Cultural Heritage Biodeterioration Studies
So far, the Oxford Nanopore® MinION™ sequencer has been applied in a variety of cultural heritage materials including: stone monuments, granite chapels, salt-weathered buildings, petroglyph sites, oil paintings, drawings, textiles, waxes, bronze statues, waterlogged archeological wood pirogues, iron nails from a whale skeleton, documents and in museum environments (Table 1).
Grottoli and colleagues [18] used a gene sequencing tailored approach (ITS, 16S and 18S rDNA regions) to apply the Oxford Nanopore® MinION™ sequencer in a study in the hypogeum of Basilica di San Nicola, in Carcere Church (Rome, Italy). The authors developed and tested a bioinformatics approach, named “AmpLIcon SequencIng Analysis” (ALISIA) to evaluate their results, and verified a feeble overlap between this approach and culture dependent methods. Kisová and colleagues [19] used a metabarcoding approach (ITS, 16S and 28S rDNA regions) to study the microbiomes of funeral accessories. The authors were able to characterize biodeteriorative microorganisms, including bacteria responsible for metal corrosion and bio-mineralization, and entomopathogenic and phytopathogenic fungi problematic in these textiles. Šoltys and colleagues [20] sequenced the ITS, 16S and 28S rDNA regions to study the biodeteriogenic microbiome of a XVIII Century wax seal colored with minium. The authors identified the presence of a complex microbiota dominated by fungi capable of conducting alterations to lipids, leads, and contributing to the formation of lead soaps and secondary biogenic minerals. Piñar and colleagues [21,22] conducted two studies using a whole genome amplification (WGA) approach in various oil paintings and Leonardo da Vinci’s drawings. The authors were able to establish a fast and simple molecular WGA protocol for application in the area, while also highlighting microbiome variances in different composition and conservation status of distinct paintings [21]. For the Leonardo da Vinci’s drawings [22], they were able to observe the impacts of geographical region on the microbiome and obtain a microbiological “bio-archive”, and consequently a reference dataset for future monitoring efforts. Planý and colleagues [23] conducted a metabarcoding study of the 16S and 28S rDNA regions to understand iron nails corrosion from a whale skeleton displayed at the Natural History Museum in London, United Kingdom. Through this method, they were able to characterize the fungal and bacterial communities associated with the corrosion of the iron nails, while also proposing a fungal-mediated biodeterioration mechanism. Brimblecombe and colleagues [24] sequenced the ITS region to study the fungal surface contamination in the Klosterneuburg Monastic Library. The authors also compared these results with cultivated fungi retrieved from library dust at this site. Delegou and colleagues [25] applied 16S rDNA sequencing to characterize the microbiota of building materials of the holy Aedicule sepulcher in Jerusalem. They were also able to characterize and discuss the biodeteriorative impact of the major species found. Pavlović and colleagues [26] analyzed various genes (ITS and 16S rDNA region; and the nitrite reductase (nirK), sulfite reductase (dsr), the sox enzyme system for sulfur oxidation (soxB) and the ammonia monooxygenase (amoA) genes) to study a salt-contaminated twelfth century granite-built chapel. The authors contributed to the knowledge of the role of microorganisms in the presence of salt and damp stains, by founding a well-defined relationship between the microbiome and soluble salts, and consequently the biodeterioration phenomena affecting these chapels. Through the analysis of the ITS, 16S and 28S rDNA regions, Pavlović and colleagues [27] studied the microbiome of wax drippings from a candle on manuscripts. They found that wax did not make the paper more biodegradable, but provided niches for the accumulation of dust and eroded material, which in turn could act as nutritional hotspots for microbial proliferation. Rabbachin and colleagues [28] performed WGA sequencing in petroglyph sites of the Negev desert of Israel. The authors corroborated the connections of the microbial communities between rock varnish and stone inhabitants, and the microbiome contribution to the deterioration visualized. Timoncini and colleagues [29] conducted 16S rDNA sequencing to understand the role of microbial communities on bronze and marble statues patinas. They verified that differential bacterial communities occurred between marble and bronze statues and among different marble patinas. Additionally, the authors noted high microbial diversities in marble surfaces, low microorganism diversity in bronze statues, being able to expand the current knowledge regarding bacteria communities and various patinas. Beccaccioli and colleagues [30] targeted the ITS and 16S rDNA regions to understand the microbial communities contributing to the deterioration of waterlogged wooden fragments from pirogues. The authors also developed and tested a custom pipeline for their bioinformatics analysis. The results obtained allowed to identify high levels of many bacteria and ligninolytic fungi able to contribute to wood erosion of these artifacts. Li and colleagues [31] used a WGA approach (Illumina + Nanopore) in biofilms thriving in Leshan and Feilaifeng stone cultural heritage sites. They were able to understand the biofilms’ taxonomic profiles, while also gathering a strong knowledge of the microbial groups and gene families contributing to elemental nitrogen and sulfur metabolism enhancing stone biodeterioration. In addition, the authors were also able to shed light on biofilm resistome (see below). Nir and colleagues [32] applied the Oxford Nanopore® MinION™ sequencer to gather knowledge on the genomic characteristics of cyanobacteria colonizing the Negev petroglyphs. The authors were able to generate a metagenome from Trichocoleus desertorum strain NBK24 to understand genes crucial for survival at these extreme environments and contributing to elemental exchanges leading to biodeterioration. Pavlović and colleagues [33] conducted a metabarcoding analysis of 16S and 28S rDNA regions to study the microbiome of books, folklore prints and archive documents. The authors noted that complex microbial communities were responsible to stain two types of paper analyzed. Rabbachin and colleagues [34] applied metagenomic sequencing (WGA) to study natural patinas in petroglyphs in the Austrian Alps. They found differences on stone microbiomes with and without visible biofilm/patinas and concluded that their removal could enhance and continue new cycles of colonization and biodeterioration. Through the metabarcoding analysis of the 16S rDNA region, Tichy and colleagues [35] were able to understand the relationships between the microbiome and pinkish patinas in salt-weathered buildings. The authors found that the microbial communities were influenced by salt concentration and chemical composition. Moreover, this study also highlighted the impact of presence/absence of K+ ions on the bacteria and archaea communities and consequently biodeterioration at these sites. Bastholm and colleagues [36] sequenced the calmodulin gene through metabarcoding approaches to identify xerophilic Aspergillus growth in Danish Museum repositories. Comparisons with Illumina amplicon sequencing and cultivation-dependent methods were also performed, in an effort to understand if Aspergillus section Restricti species had become a novel contaminant nationally distributed. Haedar and colleagues [37] conducted analysis of the 16S rDNA region to understand the bacterial communities on degraded Prehistoric rock paintings in Maros-Pangkep Global Geopark. They found multiple genera known to be involved in carbonate precipitation and rock weathering contributing to the problems noted at this site. Rabbachin and colleagues [38] examined the fungal diversity at petroglyph sites in the Negev Desert, Israel. They compared mycobiomes from stone, soil and air, discovering a high fungal biodiversity in the soil, a dominance of Cladosporium in the air and the presence of lichens and extremotolerant microcolonial fungi in the stone materials.
Overall and taking into account these works, it is clear that the Oxford Nanopore® sequencer is increasingly becoming a critical component of microbial cultural heritage biodeterioration and conservation studies [8,9,17,39]. Some trends in the application of this system can also be verified. The number of studies using a metabarcoding approach are predominant, with the most sequenced genes being the ITS, 16S and 28S rDNA regions. In parallel, the most studied supports are related to stone substrates (Figure 2). The most common keywords found in these works include: biodeterioration, metagenomics, nanopore sequencing technology, building materials, stone, microbial community, 16s RNA ribosomal gene, microbiome, bacteria, nanopore sequencing technology, MinION™, microbiota and conservation (Figure 3). Additionally, through the analysis of the most common words found in the abstracts of these manuscripts (Figure 4), some of the Oxford Nanopore® MinION™ additional tendencies can also be further understood. In fact, a great deal of focus of these works is given to microorganism abilities to grow in museum environments and drawings, the biodeterioration of stone substrates affected by salts and the resistance mechanisms of biodeteriogens.
As such, some consequently current gaps of the application of the technique in this area include the need: (1) to expand the number of studies in supports less studied or not yet explored; (2) to conduct further discussions and comparisons regarding bioinformatic pipelines and analysis; and (3) to proceed with the expansion of application of WGA tailored studies, in order to deepen the knowledge of metabolic and resistance characteristics of microorganisms. Moreover, other additional topics could also be further investigated, such as the case of biodeteriogens metatranscriptomics, microbial ecology, on-site real-time application and molecular monitoring; biotechnology focusing in preservation/restoration efforts; and the monitoring of treatment efficiency through time.
3. Future Directions for Oxford Nanopore® Applications in Biodeterioration Studies
Pavlović and colleagues [17] provided an initial review of the first five articles where the technique was applied in this area [18,19,20,21,22]. The authors also discussed that future works could focus on the study of cultural heritage microbiomes metabolic and degradative features, the further creation of biobanks, the impacts of climate change on microbes, bioinformatics difficulties and available pipelines, and the improvements being made to the device sequencing capacities.
However, during the last five years, the technique application in various differential contexts has also opened the doors for their putative usage on other interesting sub-areas of research. These include, for instance: biodeteriogens genome and transcriptome analysis, metatranscriptomics, biodeteriorative metabolism studies, inter and intra kingdom interactions analysis, resistome understanding, biocodicology, object history and context, in situ applications, bioprospecting and biotechnology (Figure 5).
3.1. Biodeteriogens Genomics and Transcriptomics
Genome-wide and transcriptomic analysis can provide valuable information regarding the genetic basis and microbial mechanisms involved in biodeterioration. Nonetheless, one of the current major challenges for advancements in this area is the lack of sequenced microbial genomes and the absence of transcriptomic experimental data. For example, for black fungi (one of the major problematic microorganisms in stone), only a very low number of genomes from isolates retrieved from stone monuments are currently available [40,41]. Additionally, the number of available transcriptomic studies in the area, is much lower.
In a recent study, Quach and colleagues [42] demonstrated the utility of genome sequencing to understand lifestyle adaptations and glass biodeterioration mechanisms of Curvularia eragrostidis with the Illumina technique. The authors were able to characterize the metabolic pathways involved in adaptations to the species glassy habitat and the genomic basis for organic acid, exopolysaccharide biosynthesis and enzymatic production (products able to conduct biodeterioration).
Paiva and colleagues [43] also used Illumina to sequence the genome of the microcolonial black fungus Saxispiralis lemnorum and conducted a deep genomic analysis of the family Aeminiaceae. The authors found, not only various peculiar features for extremotolerance, but also metabolic properties that can putatively contribute to biodeterioration, such as enzymatic characteristics and their abilities for nitrate assimilation and sulfate reduction.
Pei and colleagues [44] conducted studies on the genomic and transcriptomic characteristics of Naumannella cuiyingiana AFT2T contributing to the lead-containing pigment discoloration of a 1500 years tomb wall painting of China. The authors found that the Pb ions presence in culture media induced changes in gene expression related to metal ion transport and metabolism genes, and to TlpA family protein disulfide reductases. Enzymes with disulfide-bond reducing properties and genes encoding the divalent Pb ion uptake transporter were also highlighted as the key genes in the process of discoloration of this tomb wall.
Wang and colleagues [45] studied the biodegradation mechanism of Fusarium solani NK-NH1 on the hull wood of the Nanhai No. 1 shipwreck. The authors performed whole genome sequencing of this isolate and were able to characterize the key genes responsible for cellulose and lignin degradation.
Presently, the Oxford Nanopore® technique has been shown to be able to sequence complete bacterial and plasmid genomes without the need for short-read sequencing [46]. Complementarily, the method has also been shown to be able to obtain near-complete, telomere to telomere fungal genomes [47,48,49] and with the current advancements in flow cell technology, will surely allow for improved results in a near future. Nonetheless, neither biodeteriogenic microorganisms genomic or transcriptomic analyses have been conducted with this system in the area. Such approach could be an interesting and useful application for the technique in future studies, especially considering the long-read capacities of the device. Their application could potentially contribute (along with metatranscriptomics) to provide insights on one of the major questions of the area “What do organisms do on and with the object?” [8].
3.2. Metatranscriptomics
While a large number of studies in the area using metagenomics or metabarcoding are available, there is still a lack of metatranscriptomics works focusing on cultural heritage biodeterioration scenarios. As pointed by Sterflinger and Piñar [8], this approach could offer a huge advancement in the area since it would allow to understand the biodeteriogenic and metabolic impact of microorganisms in an object under certain conditions. Zhang and colleagues [50] have studied the active RNA-level community at the Beishiku Temple in China using the Illumina data. The authors found a congruence in the results obtained through metagenomics and metaproteomic approaches and confirmed the impact of cyanobacteria in newly formed biofilms on stone monuments at this site. However, the Oxford Nanopore® sequencer has yet to be implemented in the area for metatranscriptomics study, a topic that is highly worthy of investigations, since the sequencer has been showed to perform such types of analysis even in unusual conditions such as the ones found at the International Space Station (ISS) [51].
3.3. Biodeteriorative Metabolic Insights
While a considerable number of studies employing the sequencer have been conducted with a WGA approach (Table 1), mainly the works of Li and colleagues [32] and Nir and colleagues [33] dwelled deeper in the biodeteriorative metabolism potential from the metagenomic analysis data obtained. In the first work, this approach allowed to deeply understand metabolic pathways contributions to biogeochemical N and S cycling, known to deeply enhance stone biodeterioration [52]. The second work, focused on obtaining and characterizing the metagenome of a cyanobacteria. The results allowed to characterize genes involved in element exchange, namely ATPases and membrane transporters with potential role in weathering processes.
It is clear that even with other NGS sequencing techniques [53,54], such studies are critical for understanding not only the microbial agents responsible for biodeterioration, but also their mechanisms of action. For this reason, future studies with this sequencer in these topics can also offer a wide range of possibilities and help answer critical questions in the area (how biodeterioration occurs in each specific setting).
3.4. Multi-Kingdom Interactions and Microbial Ecology
Biodeterioration of cultural heritage materials can occasionally occur due to the impact of one or a restricted group of microorganisms, although the majority of these processes are a result from a complex microbial community that establishes and displays intra- and inter-species associations and interactions [55,56]. Indeed, multi-kingdom mutualistic, competitive and neutral relationships can shape the cultural heritage materials microbiome [55,56], and consequently the biodeterioration processes affecting them. Thus, a deeper understanding of ecological networks can also provide fundamental knowledge on biodeteriorative microbes, contributing to predict their assemblies and help in their control [56].
Liu and colleagues [55] found that multi-kingdom interactions governed the microbiome in the subterranean Chinese Dahuting Han Dynasty Tomb. The authors found that Actinobacteria and Pseudonocardiaceae, by producing volatile geosmin, could attract Colembola that contributed to their dispersion to the tomb interior from the surrounding environment (inter-kingdom mutualism). The results also highlighted that intra-kingdom competition also occurred, as Pseudonocardiaceae thrive due to the production of cellulases and potential antimicrobial substances.
Yu and colleagues [56] conducted a metabarcoding meta-analysis of ~1000 microbiomes from cultural heritage sites in contrasting environmental conditions. The authors found that, at a global scale, bacterial communities are mainly influenced by global climate, while fungal communities’ dominance is largely explained by local habitat conditions.
In one of our recent studies in a marble statue [57], we also found some peculiar results, since multiple negative correlations were found between Bacteria and Fungi at this monument. Considering that the majority of Bacteria detected were Cyanobacteria, more positive correlations were expected, since they contribute to heterotrophs development.
Considering the importance and need to understand intra- and inter-species ecological interactions to improve long-term conservation of cultural heritage materials, future studies with the Oxford Nanopore® sequencer (using metagenomics and metatranscriptomics simultaneously, for instance) could provide critical knowledge to understand both the microbiome and their metabolic traits contributing to these interactions.
3.5. Cultural Heritage Resistome
The cultural heritage resistome, i.e., the antimicrobial resistance genes (AMR or ARGs) of a community, is a topic gaining some recent focus on the area [31,58,59]. Studies on AMR genes on cultural heritage monuments and relics can be considered of great relevance, considering that they can allow to understand anthropogenic activity impacts, to monitor resistance to preservation/conservation efforts, to understand contamination source tracking and even to contribute to public health management [31,58,59].
Li and colleagues [31] used both the Illumina and the Oxford Nanopore® techniques to understand the microbiome and key genetic contexts from Leshan and Feilaifeng stone heritages. The authors applied the Oxford Nanopore® sequencer for the first time in this context, and found high abundances and diversity of antibacterial biocide and metal resistance genes to copper and quaternary ammonium compounds. Furthermore, they also verified that diverse antimicrobial resistance genes and mobile genetic elements can be transmitted through horizontal gene transfer, putatively difficulting treatment applications on these relics. A comparison between both methods is also provided by the authors, confirming the ability to: (1) obtain average longer read lengths (8510 bp vs 304,680 bp from Illumina); (2) improved contig results (9390 vs 542,843 bp from Illumina) with the Oxford Nanopore® sequencer; and (3) an overall higher quality average contig N50 value, than when solely applying the Illumina reads [31].
He and colleagues [58] studied the environmental resistome and mobilome from Feilaifeng stone heritage sites in Hangzhou, Zhejiang Province in China, with the Illumina technique. The authors found that abundant and diverse AMR genes confer resistance to drugs (antibiotics), biocides and metals in the substrates studied, and that anthropogenic activities likely impact stone resistomes across stone heritage areas. Moreover, the authors also found that stone resistome and mobilome can improve their adaptation and confer resistance to antimicrobials applied against biodeterioration.
Ding and colleagues [59] studied pathogenicity characteristics and resistome profiles of the microbiome on the Angkor sandstone monuments in Cambodia with the Illumina technique. The authors found a specific set of ARGs with cross-niches between the environment and warm blood fauna. Additionally, they also verified resistance mechanisms on these supports and discussed their implications for public health on tourism of cultural heritage sites.
Considering the importance and need to understand resistance mechanisms of treatments applied on the conservation of cultural heritage materials and the AMR treat to public health, more studies with this unique sequencer could provide a deeper knowledge for the safety of visitors, contributing towards the one-health perspective and to improve conservation efforts. Recently, the technique alone was proven to be a quick, easy and reliable method for their detection [60], and their application on the area will surely have a further deep impact of our understanding of the microbiome dynamics and resistance by monitoring treatment effectivity.
3.6. Cultural Heritage Objects History
Molecular studies have also been shown to provide important information regarding the history, context, manufacturing materials, storage conditions, geographical origin and tampering of art objects [21,22,61,62]. In addition, they can also provide important clues about interactions of the relics with human interactions [61]. As such, they can provide important information in fields such as biocodicology, archeology, criminology but also in human contamination from handling [21,22,62,63,64]. While an emerging topic, attempts to apply the Oxford Nanopore® sequencer for the identification of the microbiome and animal ancient DNA (aDNA) from a 15th-century parchment from the Graphic Collection (Kupferstichkabinett) of the Academy of Fine Arts, in Vienna [62], have also been conducted. On the other hand, their application can also enhance our understanding of the object context and to help shedding light on the biodeteriorative/bioprotective dualistic effect/nature of the microbiome [64]. Understandably, this sub-area can also open doors for multiple applications of the Oxford Nanopore® sequencer worthy of further research.
3.7. On-Site Application
Due to the sequencer portability, handling features, adaptability and its analysis speed, the platform allows for an on-site analysis. In fact, multiple reports of such applications are currently available and the technique has been applied for real-time sequencing in differential environmental contexts, such as Ebola surveillance in West Africa, water quality monitoring, environmental microbe analysis, coronavirus SARS-CoV-2 diagnosis and even aboard the ISS [65,66,67,68,69,70,71,72,73]. The technique has also demonstrated to be reliable in resource-limited settings [71], a condition that is also verified for some cultural heritage monuments worldwide. In addition, Oxford Nanopore® WGS analysis can also contribute to developing countries study of their historical properties’ microbial threats. However, the on-site capabilities have yet to be explored in this research area.
Recently, Tamames and colleagues [73] developed and tested in volcanic rocks in La Palma, Canary Islands and marine waters, an in-situ system for Oxford Nanopore® metagenomic analysis in less than one day. Understandably, with such progress, the application of the technique for on-site analysis can offer a wide range of possibilities in the cultural heritage biodeterioration area, such as the monitoring of microbial outbreaks and in the implementation of quick control measures [74,75].
Complementarily, while culture-dependent methodologies face limitations to provide a holistic perspective of the microbiome causing biodeterioration of an artifact, they can offer important information since they allow microbe isolation for differential analysis, novel microorganisms discovery and the improvement of biological databases [76,77]. The sequencer has also been shown to be effective for in situ bioprospecting applications and microbial characterization [78], which considering the often-uncharacterized components of heritage materials microbiome [77], could also be worthy of future investigations.
3.8. Biotechnology and Restoration Efforts
From a biotechnological perspective, green intervention methodologies such as bioconsolidation and biocleaning [77] could also benefit of their coupling with the sequencer. Bioconsolidation refers to the set of methodologies consisting in the application of bacteria to remediate building materials, by mimicry of bacterial precipitation of carbonates on carbonate rocks [80,81]. On the other hand, biocleaning, consists in the exploration of microbial metabolism properties that act on removal of sulphates, nitrates and organic matter, in order to clean or ameliorate affected stone surfaces [82]. In this topic, the Oxford Nanopore® sequencer could be used to explore and to further understand bioconsolidation and biocleaning mechanisms through genomic and transcriptomic analysis which in turn could open doors for genomic manipulation and enhancement of these properties. Additionally, contributions towards the possible application and improvement of alternative microbial derived strategies such as the application of antagonistic microorganisms or even bacteria able conduct to bioremoval, could also benefit from this system [83,84,85,86]. Lastly, application of the method to understand the effectiveness of biocide treatments can also be a promising outlook, as it has already been demonstrated to be useful with the Illumina system [87].
4. Challenges in the Oxford Nanopore® Application in Biodeterioration Studies
While the sequencer presents numerous advantages for studying microbial communities in cultural heritage materials, several limitations must also be acknowledged. Among them, it should be highlighted that the relative high costs for some researchers, the difficulties to attain proper reagent storage in low-resource or remote settings and the need for strong internet connections for analysis, can hamper new applications of the method [71].
On the other hand, the occurrence of sequencing errors and the complexity of bioinformatics pipelines required for data analysis can also present further challenges for researchers sing this technique. Currently, while not error-free, much of the long-read associated issues have largely been solved due to the development of multiple error correction software’s in the last years [88] and to the creation of new flow cells. In fact, the Oxford Nanopore® systems are one of the fastest developing NGS platforms today and their limitations are systematically being improved. For instance, the new flow cell R10.4.1 allows quality scores of Q20+, very high accuracy levels (often over 99%) and even allows Short Fragmented Mode (SFM) applications [62,89,90,91,92] which, in itself, can offer another additional range of possibilities.
Less consensus occurs, however, when dealing with the bioinformatic analysis of the data obtained. Current limitations and criticisms include: the need for user-friendly bioinformatics platforms; the large amount of data obtained that can lead to enormous IT resource demands [72]; and the availability of methods and comprehensive databases for these analyses [87]. Moreover, the system standard bioinformatic platforms such as Epi2me requires internet connection, although offline applications are currently also available [93]. Nonetheless, current testing of alternative bioinformatic pipelines and databases in and outside of the CHB area [18,30,33,94,95,96,97,98,99,100,101] is an ongoing effort, which will surely require additional analysis and standardization in the near future.
Additionally, while MinION™ offers portability and in-field analysis capabilities, sample preparation protocols may need optimization to ensure reproducibility and reliable results in non-laboratory settings. On the other hand, and to a lesser degree, difficulties to conduct routinary procedures in situ, such as low DNA recovery from environmental samples, also needs to be noted [73]. While protocols for improved DNA extraction (e.g., [102]) have been developed, they need to be further tested in cultural heritage biodeterioration scenarios.
Overall, addressing these limitations will understandably be crucial to effectively ensure the utility of nanopore sequencing [88] in cultural heritage biodeterioration studies and in advancing our understanding of microbial interactions and degradation processes affecting cultural heritage artifacts.
5. Conclusions
So far, the application of Oxford Nanopore® MinION™ sequencing technology in cultural heritage biodeterioration studies has demonstrated a high potential and versatility. The technology has been effectively been applied in a wide range of materials, allowing the study of the diverse microbial communities and some metabolic pathways involved in their biodeterioration. Current trends in the area, include a tendency towards gene amplification tailored studies and a great focus in stone related materials. Future perspectives for the application of the technique in this field are promising, since due to the sequencer characteristics, they can be applied: for in situ analysis and bioprospecting; to microorganisms deeper genomic, transcriptomic and environmental metatranscriptomics analysis to understand active metabolic processes; to understand inter- and intra-kingdom microbial interactions and to understand objects history. Additionally, exploration of cultural heritage materials resistome can provide critical information on antimicrobial resistance, leading to the development and implement of improved conservation strategies. Furthermore, the technology's use in biotechnology could allow to the enhancement of innovative conservation methods. In conclusion, the sequencer has become an invaluable tool in the field of cultural heritage conservation and the exploration of new applications in the area can contribute to the preservation of cultural heritage relics and sites for future generations.
Author Contributions
Conceptualization, J.T.; formal analysis, J.T. and F.S.; investigation, J.T. and F.S.; writing—original draft preparation, J.T.; writing—review and editing, F.S.; project administration, J.T. and F.S.; funding acquisition, J.T.; All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Santander/University of Coimbra in the framework of the SeedProjects@UC 2024 with the reference PT0054.A.01.C. This work was carried out at the R&D Unit Centre for Functional Ecology—Science for People & the Planet (CFE) and Associate Laboratory TERRA. The Centre for Functional Ecology—Science for People and the Planet (CFE) was supported by FCT - Fundação para a Ciência e Tecnologia, I.P. by project reference UIDB/04004/2020 and the DOI identifier 10.54499/UIDB/04004/2020 (https://doi.org/10.54499/UIDB/04004/2020). The Associate Laboratory TERRA was supported by FCT - Fundação para a Ciência e Tecnologia, I.P. by project reference LA/P/0092/2020 and the DOI identifier 10.54499/LA/P/0092/2020 (https://doi.org/10.54499/LA/P/0092/2020). The authors also thank the funding of PRR—Recovery and Resilience Plan and by the NextGeneration EU European Funds.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hueck, H. J. The biodeterioration of materials as part of hylobiology. Mater Org, 1965, 1, 1. 5– 34.
- Hueck, H.J. , The biodeterioration of materials – an appraisal. In Biodeterioration of Materials. Walters, A.H., Elphick, J.S., Eds, Elsevier, London, UK, 1968, 6–12.
- Urzì, C. , Krumbein. W.E., Microbiological impacts on the cultural heritage. In Durability and change: the science, responsability and cost of sustaining cultural heritage. Krumbein, W.E., Brimblecombe, P., Cosgrove D.E., Stainforth, S., Eds, John Wiley & Sons Ltd., London, UK, 1994, 107–135.
- Sterflinger, K.; Piñar, G. Microbial Deterioration of Cultural Heritage and Works of Art--Tilting at Windmills? Appl Microbiol Biotechnol. 2013, 97, 9637–9646. [Google Scholar] [CrossRef] [PubMed]
- Favero-Longo, S.E.; Viles, H.A. A Review of the Nature, Role and Control of Lithobionts on Stone Cultural Heritage: Weighing-up and Managing Biodeterioration and Bioprotection. World J. Microbiol. Biotechnol. 2020, 36, 100. [Google Scholar] [CrossRef]
- Liu, X.; Qian, Y.; Wu, F.; Wang, Y.; Wang, W.; Gu, J.-D. Biofilms on Stone Monuments: Biodeterioration or Bioprotection? Trends Microbiol. 2022, 30, 816–819. [Google Scholar] [CrossRef]
- Beata, G. The Use of -Omics Tools for Assessing Biodeterioration of Cultural Heritage: A Review. J Cult Herit. 2020, 45, 351–361. [Google Scholar] [CrossRef]
- Sterflinger, K.; Piñar, G. Molecular-Based Techniques for the Study of Microbial Communities in Artworks. In Microorganisms in the Deterioration and Preservation of Cultural Heritage; Joseph, E., Ed.; Springer International Publishing: Cham. 2021, 59–77 ISBN 978-3-030-69411-1.
- Piñar, G.; Sterflinger, K. Natural Sciences at the Service of Art and Cultural Heritage: An Interdisciplinary Area in Development and Important Challenges. Microb Biotechnol. 2021, 14, 806–809. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.J.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim Biophys Acta. 2014, 1842, 1932–1941. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat Rev Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Senbeta, B.; Worku, T.; Mohammed, A.A.; Senbeta, B.; Worku, T. Pacific bioscience sequence technology: Review. Int J Vet Sci Res. 2022, 8, 27–33. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Pavlovic, J.; Cavalieri, D.; Mastromei, G.; Pangallo, D.; Perito, B.; Marvasi, M. MinION Technology for Microbiome Sequencing Applications for the Conservation of Cultural Heritage. Microbiol Res. 2021, 247, 126727. [Google Scholar] [CrossRef]
- Grottoli, A.; Beccaccioli, M.; Zoppis, E.; Fratini, R.S.; Schifano, E.; Santarelli, M.L.; Uccelletti, D.; Reverberi, M. Nanopore Sequencing and Bioinformatics for Rapidly Identifying Cultural Heritage Spoilage Microorganisms. Front Mater. 2020, 7. [Google Scholar] [CrossRef]
- Kisová, Z.; Planý, M.; Pavlović, J.; Bučková, M.; Puškárová, A.; Kraková, L.; Kapustová, M.; Pangallo, D.; Šoltys, K. Biodeteriogens Characterization and Molecular Analyses of Diverse Funeral Accessories from XVII Century. Appl Sci. 2020, 10, 5451. [Google Scholar] [CrossRef]
- Šoltys, K.; Planý, M.; Biocca, P.; Vianello, V.; Bučková, M.; Puškárová, A.; Sclocchi, M.C.; Colaizzi, P.; Bicchieri, M.; Pangallo, D.; et al. Lead Soaps Formation and Biodiversity in a XVIII Century Wax Seal Coloured with Minium. Environ Microbiol. 2020, 22, 1517–1534. [Google Scholar] [CrossRef]
- Pinar, G.; Poyntner, C.; Lopandic, K.; Tafer, H.; Sterflinger, K. Rapid Diagnosis of Biological Colonization in Cultural Artefacts Using the MinION Nanopore Sequencing Technology. Int Biodeterior Biodegradation. 2020, 148. [Google Scholar] [CrossRef]
- Piñar, G.; Sclocchi, M.C.; Pinzari, F.; Colaizzi, P.; Graf, A.; Sebastiani, M.L.; Sterflinger, K. The Microbiome of Leonardo Da Vinci’s Drawings: A Bio-Archive of Their History. Front Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Planý, M.; Pinzari, F.; Šoltys, K.; Kraková, L.; Cornish, L.; Pangallo, D.; Jungblut, A.D.; Little, B. Fungal-Induced Atmospheric Iron Corrosion in an Indoor Environment. Int Biodeterior Biodegradation. 2021, 159, 105204. [Google Scholar] [CrossRef]
- Brimblecombe, P.; Sterflinger, K.; Derksen, K.; Haltrich, M.; Querner, P. Thermohygrometric Climate, Insects and Fungi in the Klosterneuburg Monastic Library. Heritage. 2022, 5, 4228–4244. [Google Scholar] [CrossRef]
- Delegou, E.; Karapiperis, C.; Hilioti, Z.; Chasapi, A.; Valasiadis, D.; Alexandridou, A.; Rihani, V.; Kroustalaki, M.; Bris, T.; Ouzounis, C.; et al. Metagenomics of the built cultural heritage: microbiota characterization of the building materials of the holy aedicule of the holy sepulchre in Jerusalem. 2022, CIENTIFIC CULTURE, 8(2), 59-83. [CrossRef]
- Pavlović, J.; Bosch-Roig, P.; Rusková, M.; Planý, M.; Pangallo, D.; Sanmartín, P. Long-Amplicon MinION-Based Sequencing Study in a Salt-Contaminated Twelfth Century Granite-Built Chapel. Appl Microbiol Biotechnol. 2022, 106, 4297–4314. [Google Scholar] [CrossRef]
- Pavlović, J.; Sclocchi, M.C.; Planý, M.; Ruggiero, D.; Puškárová, A.; Bučková, M.; Šoltys, K.; Colaizzi, P.; Riccardi, M.L.; Pangallo, D.; et al. The Microbiome of Candle Beeswax Drops on Ancient Manuscripts. Int Biodeterior Biodegradation. 2022, 174, 105482. [Google Scholar] [CrossRef]
- Rabbachin, L.; Piñar, G.; Nir, I.; Kushmaro, A.; Pavan, M.J.; Eitenberger, E.; Waldherr, M.; Graf, A.; Sterflinger, K. A Multi-Analytical Approach to Infer Mineral–Microbial Interactions Applied to Petroglyph Sites in the Negev Desert of Israel. Appl Sci. 2022, 12, 6936. [Google Scholar] [CrossRef]
- Timoncini, A.; Costantini, F.; Bernardi, E.; Martini, C.; Mugnai, F.; Mancuso, F.; Sassoni, E.; Ospitali, F.; Chiavari, C. Insight on Bacteria Communities in Outdoor Bronze and Marble Artefacts in a Changing Environment. Sci Total Environ. 2022, 850, 157804. [Google Scholar] [CrossRef]
- Beccaccioli, M.; Moricca, C.; Faino, L.; Reale, R.; Mineo, M.; Reverberi, M. The Neolithic Site “La Marmotta”: DNA Metabarcoding to Identify the Microbial Deterioration of Waterlogged Archeological Wood. Front Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Li, Q.; Wu, C.; He, J.; Zhang, B. Unraveling the Microbiotas and Key Genetic Contexts Identified on Stone Heritage Using Illumina and Nanopore Sequencing Platforms. Int Biodeterior Biodegradation. 2023, 185. [Google Scholar] [CrossRef]
- Nir, I.; Barak, H.; Rabbachin, L.; Arielle, K.; Pavan, M.; Winter, E.; Pinar, G.; Sterflinger, K.; Ariel, K. Trichocoleus Desertorum Isolated from Negev Desert Petroglyphs: Characterization, Adaptation and Bioerosion Potential. The Sci Total Environ. 2023, 904, 166739. [Google Scholar] [CrossRef]
- Pavlović, J.; Puškárová, A.; Planý, M.; Farkas, Z.; Rusková, M.; Kvalová, K.; Kraková, L.; Bučková, M.; Pangallo, D. Colored Stains: Microbial Survey of Cellulose-Based and Lignin Rich Papers. Int J Biol Macromol. 2023, 241, 124456. [Google Scholar] [CrossRef]
- Rabbachin, L.; Pinar, G.; Nir, I.; Kushmaro, A.; Eitenberger, E.; Waldherr, M.; Graf, A.; Sterflinger, K. Natural Biopatina on Historical Petroglyphs in the Austrian Alps: To Clean or Not to Clean? Int Biodeterior Biodegradation. 2023, 183, 105632. [Google Scholar] [CrossRef]
- Tichy, J.; Waldherr, M.; Ortbauer, M.; Graf, A.; Sipek, B.; Jembrih-Simbuerger, D.; Sterflinger, K.; Piñar, G. Pretty in Pink? Complementary Strategies for Analysing Pink Biofilms on Historical Buildings. Sci Total Environ. 2023, 904, 166737. [Google Scholar] [CrossRef]
- Bastholm, C.J.; Andersen, B.; Frisvad, J.C.; Oestergaard, S.K.; Nielsen, J.L.; Madsen, A.M.; Richter, J. A Novel Contaminant in Museums? A Cross-Sectional Study on Xerophilic Aspergillus Growth in Climate-Controlled Repositories. Sci Total Environ. 2024, 173880. [Google Scholar] [CrossRef]
- Haedar, N.; Iqram, M.; Priosambodo, D.; Lebe, R. Bacterial Communities on Degraded Prehistoric Rock Paintings in Maros-Pangkep Global Geopark. Philippine Journal of Science, 2024, 153(1): 391-402.
- Rabbachin, L.; Nir, I.; Waldherr, M.; Vassallo, Y.; Piñar, G.; Graf, A.; Kushmaro, A.; Sterflinger, K. Diversity of Fungi Associated with Petroglyph Sites in the Negev Desert, Israel, and Their Potential Role in Bioweathering. Front Fungal Biol 2024, 5. [Google Scholar] [CrossRef]
- Marvasi, M.; Pangallo, D.; Cavalieri, D.; Poyatos-Jiménez, F. Editorial: Multi-Omics Revolution in Microbial Cultural Heritage Conservation. Front Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- De Leo, F.; Marchetta, A.; Urzì, C. Black Fungi on Stone-Built Heritage: Current Knowledge and Future Outlook. Appl Sci. 2022, 12, 3969. [Google Scholar] [CrossRef]
- Trovão, J.; Tiago, I.; Soares, F.; Paiva, D.S.; Mesquita, N.; Coelho, C.; Catarino, L.; Gil, F.; Portugal, A. High-Quality Draft Genome Sequence of the Microcolonial Black Fungus Aeminium Ludgeri DSM 106916. Microbiol Resour Announc. 2019, 8, e00202–19. [Google Scholar] [CrossRef]
- Quach, N.T.; Ngo, C.C.; Nguyen, T.H.; Nguyen, P.L.; Vu, T.H.N.; Phan, T.H.T.; Nguyen, Q.H.; Le, T.T.M.; Chu, H.H.; Phi, Q.-T. Genome-Wide Comparison Deciphers Lifestyle Adaptation and Glass Biodeterioration Property of Curvularia Eragrostidis C52. Sci Rep. 2022, 12, 11411. [Google Scholar] [CrossRef]
- Paiva, D.S.; Fernandes, L.; Portugal, A.; Trovão, J. First Genome Sequence of the Microcolonial Black Fungus Saxispiralis Lemnorum MUM 23.14: Insights into the Unique Genomic Traits of the Aeminiaceae Family. Microorganisms. 2024, 12, 104. [Google Scholar] [CrossRef]
- Pei, S.; Wu, F.; Chen, Y.; Ma, W.; He, D.; Zhang, Q.; Gu, J.-D.; Wang, W.; Tian, T.; Feng, H. Mechanisms of Lead-Containing Pigment Discoloration Caused by Naumannella Cuiyingiana AFT2T Isolated from 1500 Years Tomb Wall Painting of China. Int Biodeterior Biodegradation. 2023, 185, 105689. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Y.; Li, N.; Wang, C.; Ma, K.; Huang, X.; Du, J.; Guo, H.; Pan, J. Study on Biodegradation Mechanism of Fusarium Solani NK-NH1 on the Hull Wood of the Nanhai No. 1 Shipwreck. Front Microbiol. 2024, 15, 1382653. [Google Scholar] [CrossRef]
- Zhao, W.; Zeng, W.; Pang, B.; Luo, M.; Peng, Y.; Xu, J.; Kan, B.; Li, Z.; Lu, X. Oxford Nanopore Long-Read Sequencing Enables the Generation of Complete Bacterial and Plasmid Genomes without Short-Read Sequencing. Front Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Salazar, A.N.; Gorter de Vries, A.R.; van den Broek, M.; Wijsman, M.; de la Torre Cortés, P.; Brickwedde, A.; Brouwers, N.; Daran, J.-M.G.; Abeel, T. Nanopore Sequencing Enables Near-Complete de Novo Assembly of Saccharomyces Cerevisiae Reference Strain CEN.PK113-7D. FEMS Yeast Res, 2017, 17, fox074. [CrossRef]
- McGinnis, J.L.; Giguere, D.J. High-Quality Genome Assembly of a Pestalotiopsis Fungus Using DIY-Friendly Methods 2022. [version 1; peer review: 3 approved with reservations]. F1000Research, 2022, 11:442 . [CrossRef]
- Witte, T.E.; Hicks, C.; Shoukouhi, P.; Dadej, K.; Findlay, W.; Liu, M.; Overy, D.P. Chromosome-Level Draft Genome Sequences of Three Isolates of the Toxigenic Fungus Claviceps Purpurea Showing Structural Rearrangements. Microbiol Resour Announc. 2023, 12, e00234–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, F.; Gu, J.-D.; He, K.; Fang, Z.; Liu, X.; He, D.; Ding, X.; Li, J.; Han, Z.; et al. Dominance by Cyanobacteria in the Newly Formed Biofilms on Stone Monuments under a Protective Shade at the Beishiku Temple in China. Env Res. 2024, 251, 118576. [Google Scholar] [CrossRef]
- Haveman, N.J.; Khodadad, C.L.M.; Dixit, A.R.; Louyakis, A.S.; Massa, G.D.; Venkateswaran, K.; Foster, J.S. Evaluating the Lettuce Metatranscriptome with MinION Sequencing for Future Spaceflight Food Production Applications. npj Microgravity. 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Liu, X.; Koestler, R.J.; Warscheid, T.; Katayama, Y.; Gu, J.-D. Microbial Deterioration and Sustainable Conservation of Stone Monuments and Buildings. Nat Sustain. 2020, 3, 991–1004. [Google Scholar] [CrossRef]
- Wu, F.; Ding, X.; Zhang, Y.; Gu, J.-D.; Liu, X.; Guo, Q.; Li, J.; Feng, H. Metagenomic and Metaproteomic Insights into the Microbiome and the Key Geobiochemical Potentials on the Sandstone of Rock-Hewn Beishiku Temple in Northwest China. Sci Total Environ. 2023, 893, 164616. [Google Scholar] [CrossRef]
- Qian, Z.; Li, Y.; Pratush, A.; Kan, J.; Gu, J.-D.; Peng, T.; Huang, T.; Hu, Z. A Comparative Analysis of the Microbial Communities and Functional Genes of the Nitrogen Cycling in Mangroves of China, Indian and Malaysia. Int Biodeterior Biodegradation. 2024, 190, 105767. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, X.; Jin, T.; Li, Y.; Wu, B.; Yu, D.; Yu, Z.; Su, B.; Chen, R.; Feng, Y.; et al. Multikingdom Interactions Govern the Microbiome in Subterranean Cultural Heritage Sites. PNAS. 2022, 119, e2121141119. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, J.; Chen, R.; Coleine, C.; Liu, W.; Delgado-Baquerizo, M.; Feng, Y. Unearthing the Global Patterns of Cultural Heritage Microbiome for Conservation. Int Biodeterior Biodegradation. 2024, 190, 105784. [Google Scholar] [CrossRef]
- Trovão, J.; Portugal, A. The Impact of Stone Position and Location on the Microbiome of a Marble Statue. The Microbe. 2024, 2, 100040. [Google Scholar] [CrossRef]
- He, J.; Zhang, N.; Shen, X.; Muhammad, A.; Shao, Y. Deciphering Environmental Resistome and Mobilome Risks on the Stone Monument: A Reservoir of Antimicrobial Resistance Genes. Sci Total Environ. 2022, 838, 156443. [Google Scholar] [CrossRef]
- Ding, X.; Lan, W.; Li, J.; Deng, M.; Li, Y.; Katayama, Y.; Gu, J.-D. Metagenomic Insight into the Pathogenic-Related Characteristics and Resistome Profiles within Microbiome Residing on the Angkor Sandstone Monuments in Cambodia. Sci Total Environ. 2024, 918, 170402. [Google Scholar] [CrossRef]
- Solcova, M.; Demnerova, K.; Purkrtova, S. Application of Nanopore Sequencing (MinION) for the Analysis of Bacteriome and Resistome of Bean Sprouts. Microorganisms. 2021, 9, 937. [Google Scholar] [CrossRef]
- Piñar, G.; Poyntner, C.; Tafer, H.; Sterflinger, K. A Time Travel Story: Metagenomic Analyses Decipher the Unknown Geographical Shift and the Storage History of Possibly Smuggled Antique Marble Statues. Ann Microbio,l 2019, 69, 1001–1021. [Google Scholar] [CrossRef]
- Vassallo, Y.; Waldherr, M.; Lehner, E.; Graf, A.; Cappa, F.; Hartl, A.; Schober, R.; Beccaccioli, M.; Sterflinger, K.; Piñar, G.; et al. Oxford Nanopore Technologies for Biocodicology: A Case Study on a 15th-Century Parchment - Vassallo Y., Waldherr M., Lehner E., Graf A., Cappa F., Hartl A., Schober R., Beccaccioli M., Sterflinger K., Piñar G., Reverberi M. In; DTC Lazio: Tecnologie e patrimonio culturale: nuove competenze e professioni. 2023. https://iris.uniroma1.it/handle/11573/1695400?mode=complete.
- Simon, L.M.; Flocco, C.; Burkart, F.; Methner, A.; Henke, D.; Rauer, L.; Müller, C.L.; Vogel, J.; Quaisser, C.; Overmann, J.; et al. Microbial Fingerprints Reveal Interaction between Museum Objects, Curators, and Visitors. iScience. 2023, 26, 107578. [Google Scholar] [CrossRef]
- Cao, Y.; Bowker, M.A.; Delgado-Baquerizo, M.; Xiao, B. Biocrusts Protect the Great Wall of China from Erosion. Sci Adv. 2023, 9, eadk5892. [Google Scholar] [CrossRef]
- Castro-Wallace, S.L.; Chiu, C.Y.; John, K.K.; Stahl, S.E.; Rubins, K.H.; McIntyre, A.B.R.; Dworkin, J.P.; Lupisella, M.L.; Smith, D.J.; Botkin, D.J.; et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Sci Rep. 2017, 7, 18022. [Google Scholar] [CrossRef]
- Goordial, J.; Altshuler, I.; Hindson, K.; Chan-Yam, K.; Marcolefas, E.; Whyte, L.G. In Situ Field Sequencing and Life Detection in Remote (79°26′N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Front Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Isolation and Characterization of SARS-CoV-2 from the First US COVID-19 Patient. bioRxiv, 2020, 2020.03.02.972935.
- Latorre-Pérez, A.; Pascual, J.; Porcar, M.; Vilanova, C. A Lab in the Field: Applications of Real-Time, in Situ Metagenomic Sequencing. Biology Methods and Protocols. 2020, 5, bpaa016. [Google Scholar] [CrossRef]
- Moore, S.C.; Penrice-Randal, R.; Alruwaili, M.; Dong, X.; Pullan, S.T.; Carter, D.P.; Bewley, K.; Zhao, Q.; Sun, Y.; Hartley, C.; et al. Amplicon Based MinION Sequencing of SARS-CoV-2 and Metagenomic Characterisation of Nasopharyngeal Swabs from Patients with COVID-19 bioRxiv, 2020, 2020.03.05.20032011.
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-Time, Portable Genome Sequencing for Ebola Surveillance. Nature. 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Wasswa, F.B.; Kassaza, K.; Nielsen, K.; Bazira, J. MinION Whole-Genome Sequencing in Resource-Limited Settings: Challenges and Opportunities. Curr Clin Micro Rpt. 2022, 9, 52–59. [Google Scholar] [CrossRef]
- Werner, D.; Acharya, K.; Blackburn, A.; Zan, R.; Plaimart, J.; Allen, B.; Mgana, S.M.; Sabai, S.M.; Halla, F.F.; Massawa, S.M.; et al. MinION Nanopore Sequencing Accelerates Progress towards Ubiquitous Genetics in Water Research. Water. 2022, 14, 2491. [Google Scholar] [CrossRef]
- Tamames, J.; Jiménez-Lalana, D.; Redondo, Á.; Martínez-García, S.; De Los Rios, A. In Situ Metagenomics: A Platform for Rapid Sequencing and Analysis of Metagenomes in Less than One Day. Mol Ecol Resour. 2024, 24, e13909. [Google Scholar] [CrossRef]
- Bastholm, C.J.; Madsen, A.M.; Andersen, B.; Frisvad, J.C.; Richter, J. The Mysterious Mould Outbreak - A Comprehensive Fungal Colonisation in a Climate-Controlled Museum Repository Challenges the Environmental Guidelines for Heritage Collections. J Cult Herit. 2022, 55, 78–87. [Google Scholar] [CrossRef]
- Martin-Pozas, T.; Nováková, A.; Jurado, V.; Cuezva, S.; Fernandez-Cortes, A.; Saiz-Jimenez, C.; Sanchez-Moral, S. A Second Fungal Outbreak in Castañar Cave, Spain, Discloses the Fragility of Subsurface Ecosystems. Microb Ecol. 2024, 87, 53. [Google Scholar] [CrossRef]
- Trovão, J.; Portugal, A. Current Knowledge on the Fungal Degradation Abilities Profiled through Biodeteriorative Plate Essays. Appl Sci. 2021, 11, 4196. [Google Scholar] [CrossRef]
- Pyzik, A.; Ciuchcinski, K.; Dziurzynski, M.; Dziewit, L. The Bad and the Good—Microorganisms in Cultural Heritage Environments—An Update on Biodeterioration and Biotreatment Approaches. Materials. 2021, 14, 177. [Google Scholar] [CrossRef]
- Latorre-Pérez, A.; Gimeno-Valero, H.; Tanner, K.; Pascual, J.; Vilanova, C.; Porcar, M. A Round Trip to the Desert: In Situ Nanopore Sequencing Informs Targeted Bioprospecting. Front Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Andreolli, M.; Lampis, S.; Bernardi, P.; Calò, S.; Vallini, G. Bacteria from Black Crusts on Stone Monuments Can Precipitate CaCO3 Allowing the Development of a New Bio-Consolidation Protocol for Ornamental Stone. Int Biodeterior Biodegradation. 2020, 153, 105031. [Google Scholar] [CrossRef]
- Dhami, N.K.; Reddy, M.S.; Mukherjee, A. Application of Calcifying Bacteria for Remediation of Stones and Cultural Heritages. Front Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Reddy, M.S. Biomineralization of Calcium Carbonates and Their Engineered Applications: A Review. Front Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- Cappitelli, F. Biocleaning of Cultural Heritage Surfaces. Open Conf Proc J. 2016, 7. [Google Scholar] [CrossRef]
- Ranalli, G.; Zanardini, E. Biocleaning on Cultural Heritage: New Frontiers of Microbial Biotechnologies. Journal of Applied Microbiology. 2021, 131, 583–603. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Roig, P.; Sanmartín, P. Bioremoval of Graffiti in the Context of Current Biocleaning Research. In Microorganisms in the Deterioration and Preservation of Cultural Heritage; Joseph, E., Ed.; Springer International Publishing: Cham. 2021, 175–197 ISBN 978-3-030-69411-1.
- Cattò, C.; Sanmartín, P.; Gulotta, D.; Troiano, F.; Cappitelli, F. Bioremoval of Graffiti Using Novel Commercial Strains of Bacteria. Sci Total Environ. 2021, 756, 144075. [Google Scholar] [CrossRef]
- Sanmartín, P.; Bosch-Roig, P.; Pangallo, D.; Kraková, L.; Serrano, M. Unraveling Disparate Roles of Organisms, from Plants to Bacteria, and Viruses on Built Cultural Heritage. Appl Microbiol Biotechnol, 2023, 107, 2027–2037.
- Villar-dePablo, M.; Ascaso, C.; Rodríguez-Pérez, E.; Urizal, M.; Wierzchos, J.; Pérez-Ortega, S.; de los Ríos, A. Innovative Approaches to Accurately Assess the Effectiveness of Biocide-Based Treatments to Fight Biodeterioration of Cultural Heritage Monuments. Sci. Total Environ. 2023, 897, 165318. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Rodríguez-Pérez, H.; Flores, C. Nanopore Sequencing and Its Application to the Study of Microbial Communities. CSBJ 2021, 19, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, L.J. Is Oxford Nanopore Sequencing Ready for Analyzing Complex Microbiomes? FEMS Microbiol. Ecol. 2021, 97, fiab001. [Google Scholar] [CrossRef]
- Ni, Y.; Liu, X.; Simeneh, Z.M.; Yang, M.; Li, R. Benchmarking of Nanopore R10.4 and R9.4.1 Flow Cells in Single-Cell Whole-Genome Amplification and Whole-Genome Shotgun Sequencing. Comput Struct Biotechnol J. 2023, 21, 2352–2364. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Ma, S.; Cao, J.; Liao, H.; Huang, Q.; Chen, W. The Newest Oxford Nanopore R10.4.1 Full-Length 16S rRNA Sequencing Enables the Accurate Resolution of Species-Level Microbial Community Profiling. Appl Environ Microbiol. 2023, 89, e0060523. [Google Scholar] [CrossRef]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 Long-Read Sequencing Enables the Generation of near-Finished Bacterial Genomes from Pure Cultures and Metagenomes without Short-Read or Reference Polishing. Nat Methods. 2022, 19, 823–826. [Google Scholar] [CrossRef]
- Zorz, J.; Li, C.; Chakraborty, A.; Gittins, D.A.; Surcon, T.; Morrison, N.; Bennett, R.; MacDonald, A.; Hubert, C.R.J. SituSeq: An Offline Protocol for Rapid and Remote Nanopore 16S rRNA Amplicon Sequence Analysis. ISME Commun. 2023, 3, 1–11. [Google Scholar] [CrossRef]
- Chandrakumar, I.; Gauthier, N.P.G.; Nelson, C.; Bonsall, M.B.; Locher, K.; Charles, M.; MacDonald, C.; Krajden, M.; Manges, A.R.; Chorlton, S.D. BugSplit Enables Genome-Resolved Metagenomics through Highly Accurate Taxonomic Binning of Metagenomic Assemblies. Commun Biol 2022, 5, 1–10. [Google Scholar] [CrossRef]
- Curry, K.D.; Wang, Q.; Nute, M.G.; Tyshaieva, A.; Reeves, E.; Soriano, S.; Wu, Q.; Graeber, E.; Finzer, P.; Mendling, W.; et al. Emu: Species-Level Microbial Community Profiling of Full-Length 16S rRNA Oxford Nanopore Sequencing Data. Nat Methods 2022, 19, 845–853. [Google Scholar] [CrossRef]
- Fan, J.; Huang, S.; Chorlton, S.D. BugSeq: A Highly Accurate Cloud Platform for Long-Read Metagenomic Analyses. BMC Bioinformatics 2021, 22, 160. [Google Scholar] [CrossRef]
- Jung, A.; Chorlton, S.D. BugSeq 16S: NanoCLUST with Improved Consensus Sequence Classification bioRxiv. 2021. [CrossRef]
- Petrone, J.R.; Rios Glusberger, P.; George, C.D.; Milletich, P.L.; Ahrens, A.P.; Roesch, L.F.W.; Triplett, E.W. RESCUE: A Validated Nanopore Pipeline to Classify Bacteria through Long-Read, 16S-ITS-23S rRNA Sequencing. Front. Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Planý, M.; Sitarčík, J.; Pavlović, J.; Budiš, J.; Koreňová, J.; Kuchta, T.; Pangallo, D. Evaluation of Bacterial Consortia Associated with Dairy Fermentation by Ribosomal RNA (Rrn) Operon Metabarcoding Strategy Using MinION Device. Food Bioscience 2023, 51, 102308. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, H.; Ciuffreda, L.; Flores, C. NanoCLUST: A Species-Level Analysis of 16S rRNA Nanopore Sequencing Data. Bioinformatics 2021, 37, 1600–1601. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, H.; Ciuffreda, L.; Flores, C. NanoRTax, a Real-Time Pipeline for Taxonomic and Diversity Analysis of Nanopore 16S rRNA Amplicon Sequencing Data. CSBJ 2022, 20, 5350–5354. [Google Scholar] [CrossRef]
- Maghini, D.G.; Moss, E.L.; Vance, S.E.; Bhatt, A.S. Improved High-Molecular-Weight DNA Extraction, Nanopore Sequencing and Metagenomic Assembly from the Human Gut Microbiome. Nat Protoc. 2021, 16, 458–471. [Google Scholar] [CrossRef]
Figure 1.
Example of an Oxford Nanopore® MinION™ sequencer device.
Figure 2.
Alluvial plot displaying a summary of works were Oxford Nanopore® MinION™ sequencer was applied in the cultural heritage biodeterioration field.
Figure 2.
Alluvial plot displaying a summary of works were Oxford Nanopore® MinION™ sequencer was applied in the cultural heritage biodeterioration field.
Figure 3.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main keywords (appearing at least two times) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
Figure 3.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main keywords (appearing at least two times) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
Figure 4.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main words (appearing at least two times in the abstract section) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
Figure 4.
VOSviewer (https://www.vosviewer.com/) co-occurrence network displaying the main words (appearing at least two times in the abstract section) from works regarding the application of the Oxford Nanopore® sequencer in the cultural heritage biodeterioration area.
Figure 5.
Possible future directions for the Oxford Nanopore® application in biodeterioration studies.
Figure 5.
Possible future directions for the Oxford Nanopore® application in biodeterioration studies.
Table 1.
Cultural heritage biodeterioration studies conducted with the MinION™ sequencer.
| Approach | Material | Reference |
|---|---|---|
| Gene amplification | Stone monument | [18] |
| Gene amplification | Funeral accessories textiles | [19] |
| Gene amplification | Wax seal | [20] |
| Whole genome amplification | Oil painting | [21] |
| Whole genome amplification | Drawings | [22] |
| Gene amplification | Iron nails from whale skeleton | [23] |
| Gene amplification | Museum surfaces | [24] |
| Gene amplification | Stone monument | [25] |
| Gene amplification | Granite chapel | [26] |
| Gene amplification | Beeswax drops | [27] |
| Whole genome amplification | Petroglyph sites | [28] |
| Gene amplification | Bronze and marble statues | [29] |
| Gene amplification | Waterlogged wood | [30] |
| Whole genome amplification | Stone monument | [31] |
| Whole genome amplification | Petroglyph sites | [32] |
| Gene amplification | Documents | [33] |
| Whole genome amplification | Petroglyph sites | [34] |
| Gene amplification | Salt-weathered buildings | [35] |
| Gene amplification | Air and museum surfaces | [36] |
| Gene amplification | Rock Paintings | [37] |
| Gene amplification | Petroglyph sites | [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Extraction Methods Determine the Quality of Soil Microbiota Acquisition
Zhuoxin Liu
et al.
,
2024
A Bibliographic Analysis of Biofilm Control
Yuanzhao Ding
,
2024



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated