Rotational Raman Spectroscopy: The Polarizability of The Molecule Must Be Anisotropic
Rotational Raman Spectroscopy: The Polarizability of The Molecule Must Be Anisotropic
Download as pdf or txt
You might also like
- Microwave SpectrosDocument46 pagesMicrowave Spectros5fdt78kgscNo ratings yet
- Raman 04Document43 pagesRaman 04Surender Dilip100% (1)
- Basic Principle of Cyclotron: V. S. PanditDocument9 pagesBasic Principle of Cyclotron: V. S. PanditAkshat Kumar AgarwalNo ratings yet
- X-ray Photoelectron Spectroscopy: An introduction to Principles and PracticesFrom EverandX-ray Photoelectron Spectroscopy: An introduction to Principles and PracticesNo ratings yet
- EP405Jul2021 (Part 2)Document63 pagesEP405Jul2021 (Part 2)Ayush TarwayNo ratings yet
- Raman Spectroscopy: B.Sc. (H) ChemistryDocument10 pagesRaman Spectroscopy: B.Sc. (H) ChemistrySANJAY BHAVARIYANo ratings yet
- EP405Jul2021 (IR Raman Slides)Document18 pagesEP405Jul2021 (IR Raman Slides)Ayush TarwayNo ratings yet
- Spectroscopy: Lecture 5: Application of Raman SpectrosDocument50 pagesSpectroscopy: Lecture 5: Application of Raman SpectrosMurali Krishna.HARINo ratings yet
- Organic ChemistryDocument101 pagesOrganic ChemistryHasithaNo ratings yet
- Atomic StructureDocument28 pagesAtomic StructurePavan GoudNo ratings yet
- CY1051 Raman Spectrosocpy (Uploaded)Document21 pagesCY1051 Raman Spectrosocpy (Uploaded)Valorant GlitchpopNo ratings yet
- Raman SpectrosDocument3 pagesRaman SpectroskuthappadyNo ratings yet
- PQR BranchesDocument23 pagesPQR BranchesKalpana Singh29% (7)
- Molecular SpectrosDocument51 pagesMolecular SpectrosYttrium PrasadNo ratings yet
- NMR and ESR NotesDocument38 pagesNMR and ESR NotesJasonLopez100% (1)
- Rotational Spectroscopy Part 1Document17 pagesRotational Spectroscopy Part 1shrivastavashubhang02No ratings yet
- Collective ModelDocument22 pagesCollective ModelYogendra MeenaNo ratings yet
- Unit 4Document46 pagesUnit 4Hemu chintaNo ratings yet
- NMR SpectrosDocument35 pagesNMR SpectrosSagar UpretiNo ratings yet
- Microwave Infrared: SpectrosDocument66 pagesMicrowave Infrared: SpectrosPrathamesh Dash100% (2)
- Rotational Raman SpectrosDocument17 pagesRotational Raman SpectrosAnonymous SVy8sOsvJDNo ratings yet
- Atomic Term SymbolDocument13 pagesAtomic Term SymbolUmendra KhokharNo ratings yet
- Vibrational Spectroscopy (IR, Raman)Document37 pagesVibrational Spectroscopy (IR, Raman)Destruidor 300No ratings yet
- General Chemistry: CHEM F111Document30 pagesGeneral Chemistry: CHEM F111Harsh TiwariNo ratings yet
- Introduction To Molecular SpectrosDocument13 pagesIntroduction To Molecular Spectrosapi-250366166No ratings yet
- Chapter 1 - Basic Concepts: Atoms: Discovery of Atomic Structure Rutherford (1910)Document19 pagesChapter 1 - Basic Concepts: Atoms: Discovery of Atomic Structure Rutherford (1910)Anulisa DasNo ratings yet
- Lecture 10 - SlidesDocument25 pagesLecture 10 - SlidesDeepak BhartiNo ratings yet
- Ch5 - Rotation SpectraDocument13 pagesCh5 - Rotation SpectraRajkumar MuthumanickamNo ratings yet
- Optical Spectra and X-RaysDocument19 pagesOptical Spectra and X-Rayssanjay sNo ratings yet
- Raman Scattering - Lecture 10Document11 pagesRaman Scattering - Lecture 10Omar AlshekhliNo ratings yet
- NMR MS Spectroscopy Ambo2 BDocument145 pagesNMR MS Spectroscopy Ambo2 Bkiya01No ratings yet
- Identical Particles PDFDocument31 pagesIdentical Particles PDFHồng NhânNo ratings yet
- Chapter 8 - Nuclear Magnetic Resonance SpectrosDocument38 pagesChapter 8 - Nuclear Magnetic Resonance SpectrosBizuayehu Gero100% (2)
- NMRDocument173 pagesNMRঋ ত্বিকNo ratings yet
- Chemistry 1A Spectroscopy.: Prof. Mike Ashfold (S305) (Mike - Ashfold@bris - Ac.uk)Document63 pagesChemistry 1A Spectroscopy.: Prof. Mike Ashfold (S305) (Mike - Ashfold@bris - Ac.uk)Abd El-Fattah Mohamed OufNo ratings yet
- Spectroscopy - Rotational Spectroscopy - WikiversityDocument9 pagesSpectroscopy - Rotational Spectroscopy - WikiversityDr. Gazi Jahirul ISlamNo ratings yet
- Microwave Spectroscopy BSc-Lect-2-1Document43 pagesMicrowave Spectroscopy BSc-Lect-2-1Varun JogiNo ratings yet
- Atomic SpectrosDocument36 pagesAtomic SpectrosAswin AlexNo ratings yet
- ZZ210431440 PDFDocument45 pagesZZ210431440 PDFAJER JOURNALNo ratings yet
- Chapter 1 - Basic Concepts: Atoms: Prerequisite KnowledgeDocument20 pagesChapter 1 - Basic Concepts: Atoms: Prerequisite KnowledgemahyarbNo ratings yet
- NMR 1Document60 pagesNMR 1riya100% (1)
- General Properties of Nuclei - IIIDocument8 pagesGeneral Properties of Nuclei - IIIAaush PradhanNo ratings yet
- Nuclear Magnetic Resonance (NMR) SpectrosDocument47 pagesNuclear Magnetic Resonance (NMR) SpectrosFrancisco Javier Escobar MedinaNo ratings yet
- Rovibrational SpectrosDocument9 pagesRovibrational SpectrosPrasad DurgaNo ratings yet
- By Syed Ahsan (Upload)Document12 pagesBy Syed Ahsan (Upload)Syed Ahsan Ali ShahNo ratings yet
- Microwave SpectrosDocument46 pagesMicrowave SpectrosMOHAMMED ABDUL HAINo ratings yet
- Lecture 4 Wavefunction NewDocument53 pagesLecture 4 Wavefunction NewkedirNo ratings yet
- Microwave Spectroscopy BSC Lect 2Document43 pagesMicrowave Spectroscopy BSC Lect 2anon_916856395100% (1)
- NMR Spectroscopy: by Darshan R. Telange, KNCP, Butibori (Nagpur)Document60 pagesNMR Spectroscopy: by Darshan R. Telange, KNCP, Butibori (Nagpur)team engineer100% (1)
- BITS Pilani Chemistry Hamilton OperatorDocument11 pagesBITS Pilani Chemistry Hamilton OperatorNaresh SehdevNo ratings yet
- Nuclear Magnetic Resonance (NMR) SpectrosDocument52 pagesNuclear Magnetic Resonance (NMR) Spectrossharifah sakinah syed soffianNo ratings yet
- Chapter 2Document51 pagesChapter 2Nesrine CherguiNo ratings yet
- Polarization: Dipartimento Di Fisica Universita' Della Calabria Corso Di Laurea Magistrale in Fisica A.A. 2017/2018Document9 pagesPolarization: Dipartimento Di Fisica Universita' Della Calabria Corso Di Laurea Magistrale in Fisica A.A. 2017/2018Diego Blady HaroNo ratings yet
- Raman SpectrosDocument10 pagesRaman SpectrosSruthiNo ratings yet
- AtomsDocument12 pagesAtomsmidhunesh41No ratings yet
- Bhas Mastani Summer School 2014Document81 pagesBhas Mastani Summer School 2014kashifNo ratings yet
- Hecht - Chapter 8Document47 pagesHecht - Chapter 8JunHyoung KimNo ratings yet
- 2024 - 물리화학2 - Ch 8Document53 pages2024 - 물리화학2 - Ch 8sjh585353No ratings yet
- AtomicStrucure SlidesDocument17 pagesAtomicStrucure SlidessamebalutNo ratings yet
- Spectroscopy Rotational SpectrosDocument7 pagesSpectroscopy Rotational SpectrosMONERAH D ALOSAIMINo ratings yet
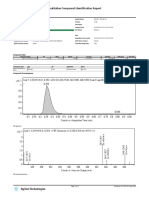
- Qualitative Compound Identification ReportDocument3 pagesQualitative Compound Identification ReportKartik RanaNo ratings yet
- 22Document1 page22Kartik RanaNo ratings yet
- Presentation (6th March, 2019)Document11 pagesPresentation (6th March, 2019)Kartik RanaNo ratings yet
- XB Literature ReportsDocument1 pageXB Literature ReportsKartik RanaNo ratings yet
- License LicDocument1 pageLicense LicKartik RanaNo ratings yet
- Sekar Research Lab: Laboratory Record NotebookDocument1 pageSekar Research Lab: Laboratory Record NotebookKartik RanaNo ratings yet
- Lectures P Block Elements 3 HypervalencyDocument26 pagesLectures P Block Elements 3 HypervalencyKartik RanaNo ratings yet
- ERT 108: Physical Chemistry: Phase DiagramsDocument36 pagesERT 108: Physical Chemistry: Phase DiagramsKartik Rana100% (1)
- Alumni Card ApplicationDocument1 pageAlumni Card ApplicationKartik RanaNo ratings yet
- Difference Between Octane and Cetane - Compare The Difference Between Similar TermsDocument7 pagesDifference Between Octane and Cetane - Compare The Difference Between Similar TermsKartik RanaNo ratings yet
- A) B) C) D) E) : Bm@iiserkol - Ac.inDocument1 pageA) B) C) D) E) : Bm@iiserkol - Ac.inKartik RanaNo ratings yet
- OJC Vol 31 (2) p605-618 PDFDocument14 pagesOJC Vol 31 (2) p605-618 PDFKartik RanaNo ratings yet
- 5.03 Optical ActivityDocument4 pages5.03 Optical ActivityKartik RanaNo ratings yet
- Closo 52 PDFDocument64 pagesCloso 52 PDFKartik RanaNo ratings yet
- C N Et - Set - Gate - Tifr: Question Bank Organometallic ChemistryDocument17 pagesC N Et - Set - Gate - Tifr: Question Bank Organometallic ChemistryKartik RanaNo ratings yet
- C N Et - Set - Gate - Tifr: Question Bank Organometallic ChemistryDocument17 pagesC N Et - Set - Gate - Tifr: Question Bank Organometallic ChemistryKartik RanaNo ratings yet
- Revista Boliviana de Química 0250-5460: IssnDocument10 pagesRevista Boliviana de Química 0250-5460: IssnKartik RanaNo ratings yet
- Organic Synthesis. ReductionsDocument64 pagesOrganic Synthesis. ReductionsKartik RanaNo ratings yet
- Homework 2Document3 pagesHomework 2Vaughan Png100% (1)
- Molpro SlidesDocument21 pagesMolpro SlidesshadrawsNo ratings yet
- Termsymbols PDFDocument4 pagesTermsymbols PDFIqbal Aljabir PujionoNo ratings yet
- Selected Solutions, Griffiths QM, Chapter 1Document4 pagesSelected Solutions, Griffiths QM, Chapter 1Kenny StephensNo ratings yet
- Physics, General Relativity: Homework Due Wednesday, NovemberDocument5 pagesPhysics, General Relativity: Homework Due Wednesday, NovemberMaci CostaNo ratings yet
- Energy Band Structure of Gold - RamchadaniDocument10 pagesEnergy Band Structure of Gold - RamchadanicarmelaNo ratings yet
- From Planck Data To Planck Era: Observational Tests of Holographic CosmologyDocument8 pagesFrom Planck Data To Planck Era: Observational Tests of Holographic CosmologyrandhyNo ratings yet
- ch36 PDFDocument11 pagesch36 PDFRodrigo S QuirinoNo ratings yet
- Quantum Gravity LecturesDocument222 pagesQuantum Gravity Lecturessidhartha samtaniNo ratings yet
- Physical Science Timeline of Atomic Models July 202020 1Document33 pagesPhysical Science Timeline of Atomic Models July 202020 1Policarpio R Princess LynNo ratings yet
- Rigid RotorDocument38 pagesRigid RotorRaniaNo ratings yet
- Atomic Model Comparison SheetDocument2 pagesAtomic Model Comparison SheetEamon BarkhordarianNo ratings yet
- Double Dot CodingDocument18 pagesDouble Dot CodingaruNo ratings yet
- Hans de Vries - Chapter 6: The Chern-Simons Electro Magnetic Spin DensityDocument22 pagesHans de Vries - Chapter 6: The Chern-Simons Electro Magnetic Spin DensityTellusz4532No ratings yet
- Stephen Hawking BiographyDocument1 pageStephen Hawking BiographyWahab AsifNo ratings yet
- Microelectronics II: EE 311A, January-May 2021Document2 pagesMicroelectronics II: EE 311A, January-May 2021Amit KumarNo ratings yet
- LS - 0 - 2 - 2d3125 - 0247b6ec34eac-Quantum ChemistryDocument2 pagesLS - 0 - 2 - 2d3125 - 0247b6ec34eac-Quantum ChemistryHamit RanaNo ratings yet
- Fluorescence and PhosphorescenceDocument15 pagesFluorescence and PhosphorescencePalupi DarmantiNo ratings yet
- Analisis de Estructuras Indeterminadas - KinnDocument12 pagesAnalisis de Estructuras Indeterminadas - KinnLucia CoxNo ratings yet
- Special Relativity - How Can Any Speed Be Defined As A Constant? - Physics Stack Exchange PDFDocument12 pagesSpecial Relativity - How Can Any Speed Be Defined As A Constant? - Physics Stack Exchange PDFKurt PatzikNo ratings yet
- Large Hadron ColliderDocument1 pageLarge Hadron ColliderMuhammad Fauzi MNo ratings yet
- The Quantum Mechanical Atom: Chemistry: The Molecular Nature of Matter, 6EDocument118 pagesThe Quantum Mechanical Atom: Chemistry: The Molecular Nature of Matter, 6Eamel andiniNo ratings yet
- HW1Document2 pagesHW1Treesha MalhotraNo ratings yet
- Special Relativity: Einstein Messes With Space and TimeDocument18 pagesSpecial Relativity: Einstein Messes With Space and TimeKýñg ButlerNo ratings yet
- Lecture 23: Introduction To Valence Bond TheoryDocument18 pagesLecture 23: Introduction To Valence Bond TheoryElectro_LiteNo ratings yet
- Introduction To Quantum Physics NotesDocument9 pagesIntroduction To Quantum Physics NotesViraj BukitagarNo ratings yet
- Note 2Document125 pagesNote 2Aiman MazlanNo ratings yet
- Identification of Compounds: Uv, Ir, NMR and Mass SpectrometriesDocument27 pagesIdentification of Compounds: Uv, Ir, NMR and Mass Spectrometries1985krNo ratings yet
- Davisson Germer LectureDocument21 pagesDavisson Germer LectureSyed Raheel AdeelNo ratings yet
- Beh H: Localized Bonding Delocalized BondingDocument33 pagesBeh H: Localized Bonding Delocalized Bondingimad IftikharNo ratings yet