0% found this document useful (0 votes)
18 viewsLecture 02
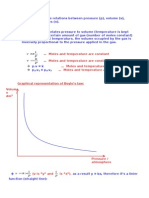
This document discusses the van der Waals equation of state, which provides a better description of gases and vapors than the ideal gas law. It describes how the van der Waals equation accounts for intermolecular forces and the finite size of molecules. The van der Waals equation predicts a critical point and negative pressures, both of which exist in nature. It also introduces the concept of corresponding states for different substances.
Uploaded by
Putu IndraCopyright
© © All Rights Reserved
Available Formats
Download as PDF, TXT or read online on Scribd
0% found this document useful (0 votes)
18 viewsLecture 02
This document discusses the van der Waals equation of state, which provides a better description of gases and vapors than the ideal gas law. It describes how the van der Waals equation accounts for intermolecular forces and the finite size of molecules. The van der Waals equation predicts a critical point and negative pressures, both of which exist in nature. It also introduces the concept of corresponding states for different substances.
Uploaded by
Putu IndraCopyright
© © All Rights Reserved
Available Formats
Download as PDF, TXT or read online on Scribd
/ 9