












































































2014 bangkok-talk
- 1. NON-INTENSIVE BIOLOGY: OPPORTUNITIES AND CHALLENGES OF NEXT-GEN SEQUENCING C. Titus Brown Assistant Professor MMG / CSE
- 2. Lansing, Michigan -> Davis, California
- 3. We practice open science! Everything discussed here: • Code: github.com/ged-lab/ ; BSD license • Blog: http://ivory.idyll.org/blog (‘titus brown blog’) • Twitter: @ctitusbrown • Grants on Lab Web site: http://ged.msu.edu/research.html • Papers available as preprints. • All my talks are available at slideshare.net/c.titus.brown/
- 4. Sequencing! • Sequencing of DNA and RNA. • Single genomes • Transcriptomes • Natural populations (tags) • Environmental samples/microbial populations (metagenomics) • Cheap and massively scalable sequencing of DNA and RNA.
- 5. Sequencing technology • Major, dramatic changes in our ability to sequence DNA and RNA quickly and cheaply. • Majority of deployed techniques depend on (variations of) a single trick: “polony” sequencing. No cloning. • Single-molecule sequencing coming along fast, but not yet ready for prime time.
- 8. Two specific concepts: • First, sequencing everything at random is very much easier than sequencing a specific gene region. (For example, it will soon be easier and cheaper to shotgun-sequence all of E. coli then it is to get a single good plasmid sequence.) • Second, if you are sequencing on a 2-D substrate (wells, or surfaces, or whatnot) then any increase in density (smaller wells, or better imaging) leads to a squared increase in the number of sequences.
- 14. Some numbers • For under $1,000 per sample, the Illumina HiSeq machine will generate: • 200,000,000 reads • Each of length ~150 • In under a week. • x 16 samples/run. • That’s almost 500 Gbp of sequence, or just over 160x human genome…
- 15. Shotgun sequencing • Collect samples; • Extract DNA or RNA; • Feed into sequencer; • Computationally analyze. “Sequence it all and let the bioinformaticians sort it Wikipedia: Environmental shotgun sequencing.png out”
- 16. The challenges of non-model sequencing • Missing or low quality genome reference. • Evolutionarily distant. • Most extant computational tools focus on model organisms – • Assume low polymorphism (internal variation) • Assume reference genome • Assume somewhat reliable functional annotation • More significant compute infrastructure …and cannot easily or directly be used on critters of interest.
- 17. Shotgun sequencing analysis goals: • Assembly (what is the text?) • Produces new genomes & transcriptomes. • Gene discovery for enzymes, drug targets, etc. • Counting (how many copies of each book?) • Measure gene expression levels, protein-DNA interactions • Variant calling (how does each edition vary?) • Discover genetic variation: genotyping, linkage studies… • Allele-specific expression analysis.
- 18. Shotgun sequencing & assembly http://eofdreams.com/library.html; http://www.theshreddingservices.com/2011/11/paper-shredding-services-small-business/; http://schoolworkhelper.net/charles-dickens%E2%80%99-tale-of-two-cities-summary-analysis/
- 19. Shotgun sequencing analysis goals: • Assembly (what is the text?) • Produces new genomes & transcriptomes. • Gene discovery for enzymes, drug targets, etc. • Counting (how many copies of each book?) • Measure gene expression levels, protein-DNA interactions • Variant calling (how does each edition vary?) • Discover genetic variation: genotyping, linkage studies… • Allele-specific expression analysis.
- 20. Assembly It was the best of times, it was the wor , it was the worst of times, it was the isdom, it was the age of foolishness mes, it was the age of wisdom, it was th It was the best of times, it was the worst of times, it was the age of wisdom, it was the age of foolishness …but for lots and lots of fragments!
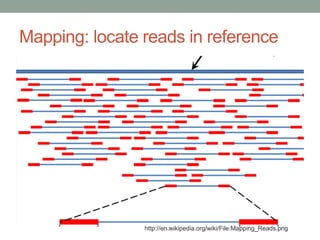
- 21. Mapping: locate reads in reference http://en.wikipedia.org/wiki/File:Mapping_Reads.png
- 22. Variant detection after mapping http://www.kenkraaijeveld.nl/genomics/bioinformatics/
- 23. Looking forward 5 years… Navin et al., 2011
- 24. Some basic math: • 1000 single cells from a tumor… • …sequenced to 40x haploid coverage with Illumina… • …yields 120 Gbp each cell… • …or 120 Tbp of data. • HiSeq X10 can do the sequencing in ~3 weeks. • The variant calling will require 2,000 CPU weeks… • …so, given ~2,000 computers, can do this all in one month.
- 25. Similar math applies: • Pathogen detection in blood; • Environmental sequencing; • Sequencing rare DNA from circulating blood. • Two issues: •Volume of data & compute infrastructure; • Latency for clinical applications.
- 26. The Data Deluge (a traditional requirement for these talks)
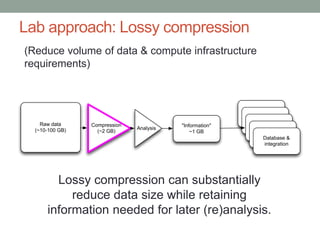
- 27. Lab approach: Lossy compression (Reduce volume of data & compute infrastructure requirements) Raw data (~10-100 GB) Analysis "Information" ~1 GB "Information" "Information" "Information" "Information" Database & integration Compression (~2 GB) Lossy compression can substantially reduce data size while retaining information needed for later (re)analysis.
- 33. Outline • The Molgulid story: investigating non-model ascidians ( this is the biology) • Meditations on data analysis. • Methods, methods, methods. •Training, training, training. • Concluding thoughts
- 34. The Molgula Story – an int’l collaboration Elijah Lowe (MSU; Naples?) Billie Swalla (UW, BEACON) Lionel Christiaen (NYU); Claudia Racioppi (Naples; NYU)
- 35. …to the urochordateswe go! Putnam et al., 2008, Modified from SwaNllaa t2u0r0e1.
- 36. Filter feeding adults Molgula oculata Molgula occulta Molgula oculata Ciona intestinalis Elijah Lowe; collaboration w/Billie Swalla
- 37. Challenging organisms to work on! Molgula occulta & M. oculata: • Only spawn ~1 month out of the year • Located off the northern coast of France • Hybrids not found outside of lab conditions • Species cannot be cultured •Wet lab techniques are not fully developed for species • No genomic resources (as of 2008).
- 38. Billie Swalla, Nadine Peyriéras, Alberto Stolfi
- 39. Tail loss and notochord a) M. oculata b) hybrid (occulta egg x oculata sperm) c) M. occulta Notochord cells in orange Swalla, B. et al. Science, Vol 274, Issue 5290, 1205-1208 , 15 November 1996
- 40. Molgula clades – tail loss is derived
- 41. Solitary ascidians have determinant and invariant cleavage. Some species have colored cytoplasms. (Boltenia villosa) The cell lineage is very similar in Ciona, Phallusia, Halocynthia roretzi & Molgula oculata.
- 42. Molgula occidentalis Ciona intestinalis
- 44. Notochord formation (convergence & extension) in ascidians is highly conserved. Ciona savignyi Jiang and Smith, 2007
- 45. Notochord Formation in Molgulids Molgula oculata notochord (40 cells, converged & extended) Molgula occulta no notochord (20 cells, not converged & extended) Hybrid notochord (20 cells, converged & extended) Swalla and Jeffery, 1996
- 46. First we applied mRNAseq… Lowe et al., in review (PeerJ). https://peerj.com/preprints/505/
- 47. …which gave us entire transcriptomes… Lowe et al., in review (PeerJ). https://peerj.com/preprints/505/
- 48. …then we sequenced their genomes... • 3 species: Molgula occidentalis (tailed) – “MOXI” Molgula oculata (tailed) – “MOCU” Molgula occulta (tail-less) – “MOCC” • 3 lanes: 300-400 bp; 650-750 bp; 900-1000 bp • ≥ 200X coverage each genome De novo assembly by Elijah Lowe (MSU) Stolfi et al., eLife, 2014; http://dx.doi.org/10.7554/eLife.03728
- 49. …which gave us most of their genes (and regulatory elements?) Genome assembly statistics: Stolfi et al., eLife, 2014; http://dx.doi.org/10.7554/eLife.03728
- 50. Shift in differentially expressed genes from gastrulation to neurulation M. ocu vs. M. occ gastrula M. ocu vs. M. occ neurula Differentially expressed during neurulation in M. ocu vs M. occ Elijah Lowe
- 51. Notochord gene expression similar to tailed species -10 -5 0 5 10 15 -10 -5 0 5 10 15 Expression difference Hybrid vs Parent species log2(hybrid)-log2(oculata) log2(hybrid)-log2(occulta) Elijah Lowe
- 52. Heterochronic Shift in MolgulidaeDevelopment *79 genes examined across six species
- 53. Transgenics of reporter constructs (“Mutual intelligibility” across ~350 my) Stolfi et al., eLife, 2014; http://dx.doi.org/10.7554/eLife.03728
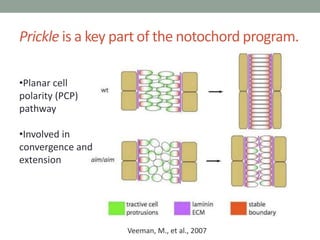
- 54. Prickle is a key part of the notochord program. Veeman, M., et al., 2007 •Planar cell polarity (PCP) pathway •Involved in convergence and extension
- 55. Prickle expressed in notochord cells of tailless ascidians. Mita et al Zool. Sci., 2010 M. occulta gastrulation Ciona intestinalis Satoh Nature Reviews Genetics 4, 2003 FGF Bra Pk Elijah Lowe
- 56. (Re)booting the Molgula -- • Determined conservation of cardiopharyngeal developmental program, despite shifts in cis-regulatory sequences (Stolfi et al, eLife, 2014). • Examining heterochronic shifts in developmental timing (tail loss) (Maliska et al., in preparation). • Connecting evolutionary shifts in developmental gene regulatory networks with conserved molecular profiles (Lowe et al, submitted; Lowe et al., in preparation).
- 57. More thoughts on Molgula • One grad student, two transcriptomes, three genomes, four years… • Genomic resources are enabling a sprawling international collaboration (UW/BEACON, MSU/BEACON, NYU, Naples, Paris) • ! Methods development key!
- 58. How Science Works Data Analysis Data generation
- 59. Luckily, data analysis is cheap and easy!
- 60. Err, well, actually… Data generation Data Analysis http://www.pixelpog.com/ftpimages/GnomesAttack.jpg
- 61. It is now easy to generate sequencing data sets of such a size and scale that the first round analysis cannot even be completed.
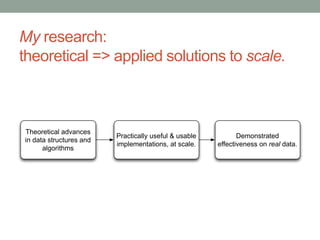
- 62. My research: theoretical => applied solutions to scale. Theoretical advances in data structures and algorithms Practically useful & usable implementations, at scale. Demonstrated effectiveness on real data.
- 63. My research: three methods. 1. Adaptation of a suite of probabilistic data structures for representing set membership and counting (Bloom filters and CountMin Sketch). (Zhang et al., PLoS One, 2014.) 2. An online streaming approach to lossy compression of sequencing data. (Brown et al., arXiv, 2012; Howe et al., PNAS, 2014.) 3. Compressible de Bruijn graph representation for assembly. (Pell et al., PNAS, 2012.)
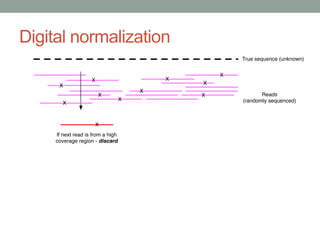
- 64. Method #2 - Digital normalization (a computational version of library normalization) Suppose you have a dilution factor of A (10) to B(1). To get 10x of B you need to get 100x of A! Overkill!! This 100x will consume disk space and, because of errors, memory. We can discard it for you…
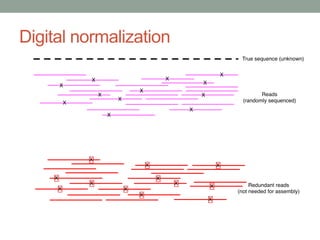
- 71. Digital normalization retains information, while discarding data and errors
- 72. Digital normalization approach A digital analog to cDNA library normalization, diginorm: • Streaming & single pass: looks at each read at most once; • Does not “collect” the majority of errors; • Keeps all low-coverage reads; • Smooths out coverage of sequencing. => Enables analyses that are otherwise completely impossible.
- 73. Witness the power of this fully operational set of sequence analysis methods: 1. Assembling soil metagenomes. Howe et al., PNAS, 2014 (w/Tiedje) 2. Understanding bone-eating worm symbionts. Goffredi et al., ISME, 2014. 3. An ultra-deep look at the lamprey transcriptome. Scott et al., in preparation (w/Li) 4. Understanding development in Molgulid ascidians. Stolfi et al, eLife 2014; etc.
- 74. Open science Guiding principle: methods that aren’t broadly available aren’t very useful. (=> Preprints, open source code, blog posts, Twitter, training, etc.) Estimated ~1000 users of our software. Diginorm now included in Trinity software from Broad Institute (~10,000 users) Illumina TruSeq long-read technology now incorporates our approach (~100,000 users)
- 75. Current research: Compressive algorithms for sequence analysis Raw data (~10-100 GB) Analysis "Information" ~1 GB "Information" "Information" "Information" "Information" Database & integration Compression (~2 GB) Can we enable and accelerate sequence-based inquiry by making all basic analysis easier and some analyses possible?
- 76. The data challenge in biology In 5-10 years, we will have nigh-infinite data. (Genomic, transcriptomic, proteomic, metabolomic, …?) We currently have no good way of querying, exploring, investigating, or mining these data sets, especially across multiple locations.. Moreover, most data is unavailable until after publication… …which, in practice, means it will be lost.
- 77. Infrastructure: distributed graph database server Web interface + API Compute server (Galaxy? Arvados?) Data/ Info Raw data sets Public servers "Walled garden" server Private server Graph query layer Upload/submit (NCBI, KBase) Import (MG-RAST, SRA, EBI)
- 78. “Data Intensive Biology” • Increasingly, relevant data is out there or can be generated fairly inexpensively. • But what does the data mean? How can we get it to yield putative answers? How can we integrate it with other people’s data? • Virtually nobody in biology is trained to do this. • Virtually nobody in biology is being trained in how to do this.
- 79. Summer NGS workshop (2010-2017)
- 80. Perspectives on training • Prediction: The single biggest challenge facing biology over the next 20 years is the lack of data analysis training (see: NIH DIWG report) • Data analysis is not turning the crank; it is an intellectual exercise on par with experimental design or paper writing. • Training is systematically undervalued in academia (!?)
- 81. Training - looking forward • NIH “Big Data 2 Knowledge” (BD2K) will be investing ~$20-40m in training each year (my estimate). Biomedical science increasingly depends on data analysis. • Moore, Sloan Foundations are investing heavily in training (see: Software Carpentry) • NSF BIO Centers have stated that “training is the second most important problem that all of us have”. We need to figure out solutions…
- 82. Funding
- 83. Students and postdocs Former: • Dr. Jason Pell (Google NYC) • Asst Professor Adina Howe (Iowa State) • Current: • Dr. Likit Preeyanon (MMG) • Elijah Lowe (CSE) • Qingpeng Zhang (CSE) • Jaron Guo (MMG) • Camille Scott (CSE) • Michael Crusoe • Luiz Irber (CSE) • Dr. Sherine Awad (MMG)
- 84. Students and postdocs Former: • Dr. Jason Pell (Google NYC) • Asst Professor Adina Howe (Iowa State) • Current: • Dr. Likit Preeyanon (MMG) • Elijah Lowe (CSE) • Qingpeng Zhang (CSE) • Jaron Guo (MMG) • Camille Scott (CSE) • Michael Crusoe • Luiz Irber (CSE) • Dr. Sherine Awad (MMG)