


















































2015 mcgill-talk
- 1. A data intensive future: how can biology best take advantage of the coming data deluge? C. Titus Brown ctbrown@ucdavis.edu Associate Professor, UC Davis
- 2. Choose your own adventure: Either you believe that all this “Big Data” stuff is nonsense and/or overblown: Please help me out by identifying my misconceptions! Or, you are interested in strategies and techniques for working with lots of data, in which case: I hope to make some useful technical and social/cultural points.
- 3. The obligatory slide about abundant sequencing data. http://www.genome.gov/sequencingcosts/ Also see: https://biomickwatson.wordpress.com/2015/03/25/the-cost-of-sequencing-is- still-going-down/
- 4. Three general uses for abundant sequencing data. Computational hypothesis falsification. Model comparison or evaluation of sufficiency. Hypothesis generation. http://ivory.idyll.org/blog/2015-what-to-do-with-sequencing-data.html
- 5. My lab’s goals re “data intensive biology” Build open tools and evaluate approaches for moving quickly from raw-ish data to hypotheses. Work with collaborators to identify emerging challenges that are preventing them from doing their science. Train peers in data analysis techniques.
- 6. Investigating soil microbial communities 95% or more of soil microbes cannot be cultured in lab. Very little transport in soil and sediment => slow mixing rates. Estimates of immense diversity: Billions of microbial cells per gram of soil. Million+ microbial species per gram of soil (Gans et al, 2005) One observed lower bound for genomic sequence complexity => 26 Gbp (Amazon Rain Forest Microbial Observatory)
- 7. N. A. Krasil'nikov, SOIL MICROORGANISMS AND HIGHER PLANTS http://www.soilandhealth.org/01aglibrary/010112krasil/010112krasil.ptII.html “By 'soil' we understand (Vil'yams, 1931) a loose surface layer of earth capable of yielding plant crops. In the physical sense the soil represents a complex disperse system consisting of three phases: solid, liquid, and gaseous.” Microbies live in & on: • Surfaces of aggregate particles; • Pores within microaggregates;
- 8. Questions to address Role of soil microbes in nutrient cycling: How does agricultural soil differ from native soil? How do soil microbial communities respond to climate perturbation? Genome-level questions: What kind of strain-level heterogeneity is present in the population? What are the phage and viral populations & dynamic? What species are where, and how much is shared between different geographical locations?
- 9. Must use culture independent approaches Many reasons why you can’t or don’t want to culture: cross-feeding, niche specificity, dormancy, etc. If you want to get at underlying function, 16s analysis alone is not sufficient. Single-cell sequencing & shotgun metagenomics are two common ways to investigate complex microbial communities.
- 10. Shotgun metagenomics Collect samples; Extract DNA; Feed into sequencer; Computationally analyze. Wikipedia: Environmental shotgun sequencing.png “Sequence it all and let the bioinformaticians sort it out”
- 11. Great Prairie Grand Challenge - -SAMPLING LOCATIONS 2008
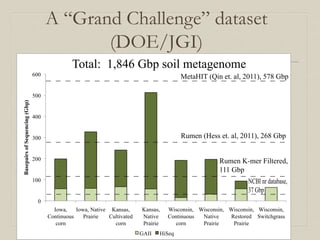
- 12. A “Grand Challenge” dataset (DOE/JGI) 0 100 200 300 400 500 600 Iowa, Continuous corn Iowa, Native Prairie Kansas, Cultivated corn Kansas, Native Prairie Wisconsin, Continuous corn Wisconsin, Native Prairie Wisconsin, Restored Prairie Wisconsin, Switchgrass BasepairsofSequencing(Gbp) GAII HiSeq Rumen (Hess et. al, 2011), 268 Gbp MetaHIT (Qin et. al, 2011), 578 Gbp NCBI nr database, 37 Gbp Total: 1,846 Gbp soil metagenome Rumen K-mer Filtered, 111 Gbp
- 13. Why do we need so much data?! 20-40x coverage is necessary; 100x is ~sufficient. Mixed population sampling => sensitivity driven by lowest abundance. For example, for E. coli in 1/1000 dilution, you would need approximately 100x coverage of a 5mb genome at 1/1000, or 500 Gbp of sequence! (For soil, estimate is 50 Tbp) Sequencing is straightforward; data analysis is not. “$1000 genome with $1m analysis”
- 14. Great Prairie Grand Challenge - goals How much of the source metagenome can we reconstruct from ~300-600 Gbp+ of shotgun sequencing? (Largest data set ever sequenced, ~2010.) What can we learn about soil from looking at the reconstructed metagenome? (See list of questions)
- 15. Great Prairie Grand Challenge - goals How much of the source metagenome can we reconstruct from ~300-600 Gbp+ of shotgun sequencing? (Largest data set ever sequenced, ~2010.) What can we learn about soil from looking at the reconstructed metagenome? (See list of questions) (For complex ecological and evolutionary systems, we’re just starting to get past the first question. More on that later.)
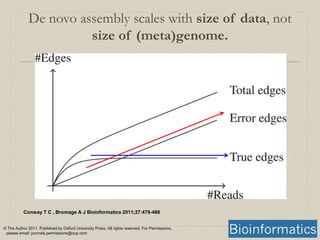
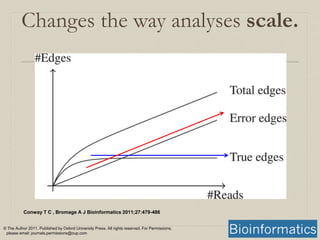
- 16. Conway T C , Bromage A J Bioinformatics 2011;27:479-486 © The Author 2011. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com De novo assembly scales with size of data, not size of (meta)genome.
- 17. Why do assemblers scale badly? Memory usage ~ “real” variation + number of errors Number of errors ~ size of data set
- 18. Our problem, in a nutshell: We had so much data that we couldn’t compute on it. (This was, and is, a common problem in non- model systems.)
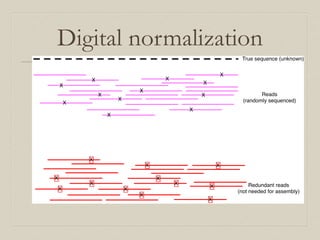
- 19. Our solution: abundance normalization (diginorm) Conway T C , Bromage A J Bioinformatics 2011;27:479-486 © The Author 2011. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com
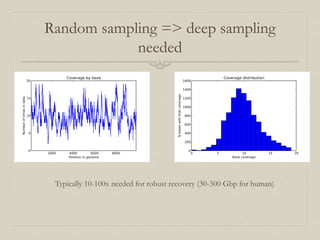
- 20. Random sampling => deep sampling needed Typically 10-100x needed for robust recovery (30-300 Gbp for human)
- 21. Actual coverage varies widely from the average. Low coverage introduces unavoidable breaks.
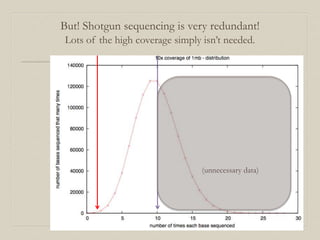
- 22. But! Shotgun sequencing is very redundant! Lots of the high coverage simply isn’t needed. (unnecessary data)
- 29. Contig assembly now scales with richness, not diversity. Most samples can be assembled on commodity computers. (information) (data)
- 30. Diginorm is widely useful: 1. Assembly of the H. contortus parasitic nematode genome, a “high polymorphism/variable coverage” problem. (Schwarz et al., 2013; pmid 23985341) 2. Reference-free assembly of the lamprey (P. marinus) transcriptome, a “big assembly” problem. (in prep) 3. Osedax symbiont metagenome, a “contaminated metagenome” problem (Goffredi et al, 2013; pmid 24225886)
- 31. Changes the way analyses scale. Conway T C , Bromage A J Bioinformatics 2011;27:479-486 © The Author 2011. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com
- 32. Question: does this approach negatively affect results? (No.) 3 70 25 1 36 13563 35 13 7 4 23 8 1 6 5 Diginorm V/O Raw V/O Diginorm trinity Raw trinity Evaluation of Molgula occulta transcriptome assembly approaches. Lowe et al., 2014, https://peerj.com/preprints/505/
- 33. Putting it in perspective: Total equivalent of ~1200 bacterial genomes Human genome ~3 billion bp Back to soil - what about the assembly results for Iowa corn and prairie?? Total Assembly Total Contigs (> 300 bp) % Reads Assembled Predicted protein coding 2.5 bill 4.5 mill 19% 5.3 mill 3.5 bill 5.9 mill 22% 6.8 mill Adina Howe
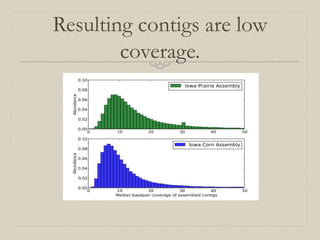
- 34. Resulting contigs are low coverage. Figure11: Coverage (median basepair) distribution of assembled contigs from soil metagenomes.
- 35. So, for soil: We really do need quite a bit more data to comprehensively sample gene content of agricultural soil; But at least now we can assemble what we already have. Estimate required sequencing depth at 50 Tbp; Now also have 2-8 Tbp from Amazon Rain Forest Microbial Observatory. …still not saturated coverage, but getting closer.
- 36. Biogeography: Iowa sample overlap? Corn and prairie De Bruijn graphs have 51% overlap. Corn Prairie Suggests that at greater depth, samples may have similar genomic content.
- 37. Putting it in perspective: Total equivalent of ~1200 bacterial genomes Human genome ~3 billion bp Blocking problem: we don’t know what most genes do! Total Assembly Total Contigs (> 300 bp) % Reads Assembled Predicted protein coding 2.5 bill 4.5 mill 19% 5.3 mill 3.5 bill 5.9 mill 22% 6.8 mill Howe et al, 2014; pmid 24632729
- 38. Reminder: the real challenge is understanding We have gotten distracted by shiny toys: sequencing!! Data!! Data is now plentiful! But: We typically have no knowledge of what > 50% of an environmental metagenome “means”, functionally. http://ivory.idyll.org/blog/2014-function-of-unknown-genes.html
- 39. Data integration as a next challenge In 5-10 years, we will have nigh-infinite data. (Genomic, transcriptomic, proteomic, metabolomic, …?) How do we explore these data sets? Registration, cross-validation, integration with models…
- 40. Carbon cycling in the ocean - “DeepDOM” cruise, Kujawinski & Longnecker et al.
- 41. Integrating many different data types to build understanding. Figure 2. Summary of challenges associated with the data integration in the proposed project. “DeepDOM” cruise: examination of dissolved organic matter & microbial metabolism vs physical parameters – potential collab.
- 43. A few thoughts on next steps. Enable scientists with better tools. Train a bioinformatics “middle class.” Accelerate science via the open science “network effect”.
- 44. That is… what now? Once you have all this data, what do you do? "Business as usual simply cannot work.” - David Haussler, 2014 Looking at millions to billions of (human) genomes in the next 5-10 years.
- 45. Enabling scientists with better tools - Build robust, flexible computational frameworks for data exploration, and make them open and remixable. Develop theory, algorithms, & software together, and train people in its use. (Oh, and stop pretending that we can develop “black boxes” that will give you the right answer.)
- 46. Education and training - towards a bioinformatics “middle class” Biology is underprepared for data-intensive investigation. We must teach and train the next generations. => Build a cohort of “data intensive biologists” who can use data and tools as an intrinsic and unremarkable part of their research. ~10-20 workshops / year, novice -> masterclass; open materials. dib-training.rtfd.org/
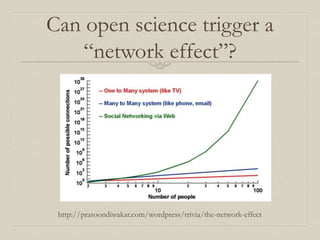
- 47. Can open science trigger a “network effect”? http://prasoondiwakar.com/wordpress/trivia/the-network-effect
- 48. The open science “network effect” If we have open tools, and trained users, then what remains to hold us back? Access to data.
- 49. The data deluge is here – it’s just somewhat hidden. I actually think this graph should be a much steeper.
- 50. Tackling data availability… In 5-10 years, we will have nigh-infinite data. (Genomic, transcriptomic, proteomic, metabolomic, …?) We currently have no good way of querying, exploring, investigating, or mining these data sets, especially across multiple locations.. Moreover, most data is unavailable until after publication, and often it must then be “curated” to become useful.
- 51. Pre-publication data sharing? There is no obvious reason to make data available prior to publication of its analysis. There is no immediate reward for doing so. Neither is there much systematized reward for doing so. (Citations and kudos feel good, but are cold comfort.) Worse, there are good reasons not to do so. If you make your data available, others can take advantage of it…
- 52. This bears some similarity to the Prisoners’ Dilemma: Where “confession” is not sharing your data. Note: I’m not a game theorist (but some of my best friends are). (Leighton Pritchard modification of http://www.acting-man.com/?p=34313)
- 53. So, how do we get academics to share their data!? Well, what are people doing now? Two successful “systems” (send me more!!) 1. Oceanographic research 2. Biomedical research
- 54. 1. Research cruises are expensive! In oceanography, individual researchers cannot afford to set up a cruise. So, they form scientific consortia. These consortia have data sharing and preprint sharing agreements. (I’m told it works pretty well (?))
- 55. 2. Some data makes more sense when you have more data Omberg et al., Nature Genetics, 2013. Sage Bionetworks et al.: Organize a consortium to generate data; Standardize data generation; Share via common platform; Store results, provenance, analysis descriptions, and source code; Run a leaderboard for a subset of analyses; Win!
- 56. This “walled garden” model is interesting! “Compete” on analysis, not on data.
- 57. Some notes - Sage model requires ~similar data in common format; Common analysis platform then becomes immediately useful; Data is ~easily re-usable by participants; Publication of data becomes straightforward; Both models are centralized and coordinated. :(
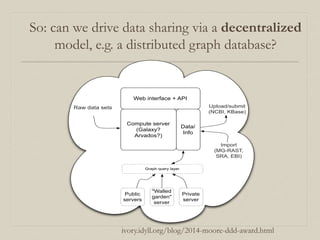
- 58. So: can we drive data sharing via a decentralized model, e.g. a distributed graph database? Compute server (Galaxy? Arvados?) Web interface + API Data/ Info Raw data sets Public servers "Walled garden" server Private server Graph query layer Upload/submit (NCBI, KBase) Import (MG-RAST, SRA, EBI) ivory.idyll.org/blog/2014-moore-ddd-award.html
- 59. My larger research vision: 100% buzzword compliantTM Enable and incentivize sharing by providing immediate utility; frictionless sharing. Permissionless innovation for e.g. new data mining approaches. Plan for poverty with federated infrastructure built on open & cloud. Solve people’s current problems, while remaining agile for the future. ivory.idyll.org/blog/2014-moore-ddd-award.html
- 60. Thanks! Please contact me at ctbrown@ucdavis.edu! Soil collaborators: Tiedje (MSU), Jansson (PNNL), Tringe (JGI/DOE)
Editor's Notes
- Fly-over country (that I live in)
- A sketch showing the relationship between the number of sequence reads and the number of edges in the graph. Because the underlying genome is fixed in size, as the number of sequence reads increases the number of edges in the graph due to the underlying genome that will plateau when every part of the genome is covered. Conversely, since errors tend to be random and more or less unique, their number scales linearly with the number of sequence reads. Once enough sequence reads are present to have enough coverage to clearly distinguish true edges (which come from the underlying genome), they will usually be outnumbered by spurious edges (which arise from errors) by a substantial factor.
- High coverage is essential.
- High coverage is essential.
- Goal is to do first stage data reduction/analysis in less time than it takes to generate the data. Compression => OLC assembly.
- Passionate about training; necessary fro advancement of field; also deeply self-interested because I find out what the real problems are. (“Some people can do assembly” is not “everyone can do assembly”)
- Analyze data in cloud; import and export important; connect to other databases.
- Work with other Moore DDD folk on the data mining aspect. Start with cross validation, move to more sophisticated in-server implementations.