























Drug development process
- 1. FACULTY OF FISHERY SCIENCES A SEMINAR ON DRUG DEVELOPMENT MECHANISMS SUBMITTED BY SOURAVBHADRA B.F.Sc 3rd yr. 1st sem ROLL NO. F/2016/29 SUBMITTED TO: PROF. Gadadhar dash H.O.D. DEPT. OF AAH
- 2. INTRODUCTION: What is Drug? Any biological,chemical,or synthetic substance which can produce biological response when it introduced into the body. What is Pharmacology? An experimenital science dealing with the properties of drug and their effects on living system. What is Pharmacokinetics & Pharmacodynamics? What the body does to the drug & What the drug does to the body.
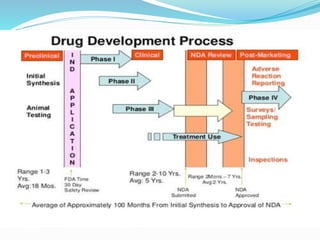
- 3. Stages of DrugDevelopment: Any drug development process must proceed through several stages in order to produce a product that is safe, efficacious, and has passed all regulatory requirements. Detailed Stages of Drug Development Discovery Product Characterization Formulation, Delivery, Packaging Development Pharmacokinetics And Drug Disposition Preclinical Toxicology Testing And IND Application Bioanalytical Testing Clinical Trials
- 4. Discovery: Typically,researchers discover new drugs through: New insights into a disease process that allow researchers to design a product to stop or reverse the effects of the disease. Many tests of moleculer compounds to find possible beneficial effects against any of a large number of disease. Existing treatments that have unanticipated effects. New technologies,such as those that provide new ways to target medical products to specific sites within the body or to manipulate genetic material.
- 5. Product Characterization: When the candidate molecule shows promise as a therapeutic, it must be characterized—the molecule’s size, shape, strengths and weaknesses, preferred conditions for maintaining function, toxicity, bioactivity, and bioavailability must be determined. Characterization studies will undergo analytical method development and validation. Early stage pharmacology studies help to characterize the underlying mechanism of action of the compound.
- 6. Formulation, Delivery, Packaging Development: Drug developers must devise a formulation that ensures the proper drug delivery parameters. Drug formulation and delivery may be refined continuously until, and even after, the drug’s final approval. Scientists determine the drug’s stability—in the formulation itself, and for all the parameters involved with storage and shipment, such as heat, light, and time. The formulation must remain potent and sterile; and it must also remain safe (nontoxic). It may also be necessary to perform leachables and extractables studies on containers or packaging.
- 7. Pharmacokinetics And Drug Disposition: Pharmacokinetic(PK)orADME(Absorption/Distribution/Met abolism/Excretion) studies provide useful feedback for formulation scientists. PK studies yield parameters such as AUC (area under the curve represents the total drug exposure across time), Cmax (maximum concentration of the drug in blood), and Tmax (time at which Cmax is reached). Later on, this data from animal PK studies is compared to data from early stage clinical trials to check the predictive power of animal models.
- 8. Preclinical Toxicology Testing and INDApplication: Preclinical testing analyzes the bioactivity, safety, and efficacy of the formulated drug product. During the preclinical stage of the development process, plans for clinical trials and an Investigational New Drug (IND) application are prepared. Studies taking place during the preclinical stage should be designed to support the clinical studies.
- 9. The main stages of preclinical toxicology testing are: Acute Toxicity Studies: Acute toxicity studies look at the effects of one or more doses administered over a period of up to 24 hours. The goal is to determine toxic dose levels and observe clinical indications of toxicity. Usually, at least two mammalian species are tested. Data from acute toxicity studies helps to determine doses for repeated dose studies in animals and Phase I studies in humans.
- 10. Repeated Dose Studies: Depending on the duration of the studies, repeated dose studies may be referred to as subacute, subchronic, or chronic. The specific duration should anticipate the length of the clinical trial that will be conducted on the new drug. Again, two species are typically required.
- 11. Genetic Toxicity Studies: These studies assess the likelihood that the drug compound is mutagenic or carcinogenic. Procedures such as the Ames test (conducted in bacteria) detect genetic changes. DNA damage is assessed in tests using mammalian cells such as the Mouse Micronucleus Test. The Chromosomal Aberration Test and similar procedures detect damage at the chromosomal level.
- 12. Reproductive Toxicity Studies Segment I reproductive toxicity studies look at the effects of the drug on fertility. Segment II and III studies detect effects on embryonic and post-natal development. In general, reproductive toxicity studies must be completed before a drug can be administered to women of child-bearing age.
- 13. Carcinogenicity Studies: . Carcinogenicity studies are usually needed only for drugs intended for chronic or recurring conditions. They are time consuming and expensive, and must be planned for early in the preclinical testing process
- 14. Bioanalytical Testing: Bioanalytical laboratory work and bioanalytical method development supports most of the other activities in the drug development process. The bioanalytical work is key to proper characterization of the molecule, assay development, developing optimal methods for cell culture or fermentation, determining process yields, and providing quality assurance and quality control for the entire development process.
- 15. Clinical Trials: Phase I Clinical Development (Human Pharmacology): Typically involves 20-80 healthy volunteers (no women of childbearing potential). Emphasis is on drug safety. Goal is to identify major side effects, metabolism and routes of excretion. Lasts about 1 year. About 70% of drugs will pass this phase.
- 16. Phase II Clinical Development (Therapeutic Exploratory): Typically involves 100-300 individuals who have the target disease. Emphasis is on effectiveness. Patients receiving the drug are compared to similar patients receiving a placebo or another drug. Lasts about 2 years. About 33% of drugs will pass this phase.
- 17. Phase III Clinical Development (Therapeutic Confirmatory): Typically involves 1000-3000 patients. Emphasis is on safety and effectiveness. Investigates through well-controlled studies different populations and different dosages as well as uses new drug in combination with other drugs. Lasts about 3 years. 25-30% of drugs will pass this phase.
- 18. Biologics License Application (BLA) or the New Drug Application (NDA): Pre-NDA period: FDA and drug sponsors meet. Submission of NDA: Formal step asking the FDA to consider approving a drug for marketing. FDA has 60 days to decide whether it will file it for approval consideration. If filed, BLAs are currently reviewed by the FDA’s Center for Biologics Evaluation and Research (CBER). NDAs are reviewed by the Center for Drug Evaluation and Research (CDER).
- 19. FDA Role: The review team evaluates the research on the safety of the drug and its effectiveness. The FDA reviews the information to go on the drug label. It inspects the facilities where the drug will be manufactured. The application will be classified as “approvable” or “not approvable”. If approvable, the FDA requests additional information from the sponsor. The NDA is again reviewed. Following drug approval, sponsors of the drug will be required to continually assess the safety of the drug.
- 20. Phase 4: Continued comparative studies. Registration and market introduction: Post-market surveillance of the drug to continually assess the safety of the drug. May include incidence and severity of rare adverse reactions, cost-effectiveness analyses, comparative trials, and quality of life studies.
- 22. RegulatoryAgency: Each country has a drug regulatory body which governs the approval process. India- CDSCO (Central Drugs Standards and Control Organization). US- FDA (Food and Drug Administration). UK- MHRA (). Medical and Healthcare Products Regulatory Agency European Union- EMEA (European Medicines Agency) Drug must be proved to be safe and effective
- 23. Conclusion: The drug discovery and development process is a long and complicated process. Before any newly discovered drug is placed on the market, it must undergo extensive testing. Each success is built on many, many prior failures. Advances in understanding human biology and disease are opening up exciting new possibilities for breakthrough medicines. At the same time, researchers face great challenges in understanding and applying these advances to the treatment of disease. These possibilities will grow as our scientific knowledge expands and becomes increasingly complex. Research-based pharmaceutical companies are committed to advancing.
- 24. References: http://www.fda.gov/Drugs/DevelopmentApprovalProc ess/default.htm https://pacificbiolabs.com/stages-of-drug- development https://en.wikipedia.org/wiki/Drug_development https://www.google.com/search?q=drug+developmen t+process+encyclopedia
- 25. THANK YOU