






























































Myelodysplasticsyndromes
- 1. MYELODYSPLASTIC SYNDROME PRESENTED BY-DR.SURENDRA GHINTALA MODERATED BY-DR. K. BHATTACHARJEE (Associate professor)) “We are put off by the fact that MDS is a heterogenous vaguely defined group of conditions with seemingly ever changing names
- 2. • First described in 1900 by Leube who used the term “Leukanamie” • Subsequently it had undergone a trial of jargon 30’s - Refractory anemia 40’s- Preleukemic anemia 50’s- Preleukemia/RARS/ Refractory normoblastic anemia 60’s-Smoldering acute leukemia 70’s –CMML/Refractory anemia with excess myeloblasts • 1982- Myelodysplastic syndrome (Benett et al )
- 3. Our bone marrow is the spongy tissue inside some of our bones, mainly flat bones. It contains immature cells, called stem cells. The stem cells can develop into the red blood cells that carry oxygen through our body, the white blood cells that fight infections, and the platelets that help with blood clotting. If we have a myelodysplastic syndrome, the stem cells do not mature into healthy blood cells. This leaves less room for healthy cells, which can lead to infection, anemia,bleeding.
- 4. • WHO definition 2008 • A group of clonal hematopoietic stem cell diseases characterized by Cytopenia Dysplasia in one or more major myeloid cell lines Ineffective hematopoiesis Increased risk of development of AML
- 5. INCIDENCE • Idiopathic MDS is a disease of the elderly; the mean age at onset is older than 70 years.( in india > 45 yrs) • There is a slight male preponderance . • MDS is a relatively common from of bone marrow failure, with reported incidence rates of 35 to >100 per million person in the general population and 120 to >500 per million in the older adult. • MDS is a rare in children, but monocytic leukemia cab be seen. • Secondary or therapy-related MDS is not age related.
- 6. The threshold for cytopenia as recommended by the IPSS for risk stratification are • Hb < 10 g/dl • Absolute neutrophil count< 1800/uL • Platelets < 1 lakh/uL
- 7. • PREDISPOSING FACTORS HEREDITARY A) Constitutional genetic disorders Downs Syndrome: 10-30 times more risk Trisomy 8 : Seen in 50% cases of MDS Monosomy 7 : Seen in 50% cases of MDS. B) Neurofibromatosis C) Congenital neutropenia syndrome Kostmann Agranulocytosis Shwachman Diamond syndrome
- 8. • D) DNA repair defects Fanconi anemia, Ataxia telangiectasia Bloom syndrome • E) Mutagen detoxification(GSTq1-null) Glutathione-S-Transferase. Studies show that GST- q1null genotype increases risk by 4 times. .
- 9. • ACQUIRED These factors play a major role in secondary MDS/ t-MDS a)Mutagen exposure 1.Genotoxic therapy- alkylating agents 2. Beta-emitter phosphorus; Used in the treatment of Polycythemia Vera- 10-15% increased risk. 3. Topoisomerase(Topo-II) interactive agents like anthracycline, etoposide. 4. Autologous stem cell transplantation- long term survivors
- 10. b) Environmental /occupational exposures Exposure to benzene-5-20 fold increase in risk. Other agents like solvents, petrochemicals,Insectide c) Tobacco Tobacco smoke contains a number of leukemogens like nitrosamines, benzene and polonium-210 d) Senescence e) Aplastic anemia
- 11. Why Classify? • Unravel disease biology • Design accurate diagnostic tests • Predict prognosis • Develop novel therapies
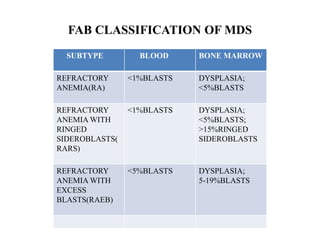
- 13. FAB CLASSIFICATION OF MDS • 1%BLAST SUBTYPE BLOOD BONE MARROW REFRACTORY ANEMIA(RA) <1%BLASTS DYSPLASIA; <5%BLASTS REFRACTORY ANEMIA WITH RINGED SIDEROBLASTS( RARS) <1%BLASTS DYSPLASIA; <5%BLASTS; >15%RINGED SIDEROBLASTS REFRACTORY ANEMIA WITH EXCESS BLASTS(RAEB) <5%BLASTS DYSPLASIA; 5-19%BLASTS
- 14. contd SUBTYPE BLOOD BONE MARROW REFRACTORY ANEMIA WITH EXCESS BLASTS IN TRANSFORMATION (RAEBt) > 5%BLASTS DYSPLASIA; 20-29%BLASTS OR AUER RODS CHRONIC MYELOMONOCYTIC LEUKEMIA(CMML) >1X109/L MONOCYTES DYSPLASIA; <30%BLASTS
- 15. WHO CLASSIFICATION OF MDS (2008) SUBTYPE BLOOD BONE MARROW REFRACTORY CYTOPENIA WITH UNILINEAGE DYSPLASIA (RCUD) REFRACTORY ANEMIA (RA), REFRACTORY NEUTROPENIA(RN) , (REFRACTORY THROMBOCYTOPENIA (RT) ANEMIA; NO OR RARE BLASTS UNICYTOPENIA BICYTOPENIA UNILINEAGE DYSPLASIA > 10% CELLS IN ONE MYELOID LINE WITH < 5% BLASTS <15%RINGED SIDEROBLASTS REFRACTORY ANEMIA WITH RINGED SIDEROBLASTS ANEMIA; NO OR RARE BLASTS >15%RINGED SIDEROBLASTS; ERYTHROID DYSPLASIA; <5%BLASTS;
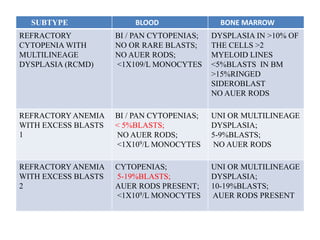
- 16. SUBTYPE BLOOD BONE MARROW REFRACTORY CYTOPENIA WITH MULTILINEAGE DYSPLASIA (RCMD) BI / PAN CYTOPENIAS; NO OR RARE BLASTS; NO AUER RODS; <1X109/L MONOCYTES DYSPLASIA IN >10% OF THE CELLS >2 MYELOID LINES <5%BLASTS IN BM >15%RINGED SIDEROBLAST NO AUER RODS REFRACTORY ANEMIA WITH EXCESS BLASTS 1 BI / PAN CYTOPENIAS; < 5%BLASTS; NO AUER RODS; <1X109/L MONOCYTES UNI OR MULTILINEAGE DYSPLASIA; 5-9%BLASTS; NO AUER RODS REFRACTORY ANEMIA WITH EXCESS BLASTS 2 CYTOPENIAS; 5-19%BLASTS; AUER RODS PRESENT; <1X109/L MONOCYTES UNI OR MULTILINEAGE DYSPLASIA; 10-19%BLASTS; AUER RODS PRESENT
- 17. SUBTYPE BLOOD BONE MARROW MYELODYSPLASTIC SYNDROME, UNCLASSIFIED(MDS-u) CYTOPENIAS; NO OR RARE BLASTS; NO AUER RODS; UNILINEAGE DYSPLASIA; <5% BLASTS; NO AUER RODS 5q-SYNDROME ANEMIA; NORMAL/INCREASED PLATELET COUNT; <5%BLASTS NORMAL/INCREASED MEGAKARYOCYTES; <5%BLASTS; NO AUER RODS CHILDHOOD MDS < 2 % BLASTS DYSPLASTIC CHANGES IN >10 % NEUTROPHILS DYSPLASTIC CHANGES IN > 10 % ERYTHROID PRECURSORS DYSPLASTIC CHANGES IN > 10 % GRANULOCYTE PRECURSORS MICROMEGAKARYOCYTES, DYSPLASTIC CHANGES IN MGKS
- 18. SUBTYPE BLOOD BONE MARROW MYELODYSPLASTIC SYNDROME, UNCLASSIFIED(MDS-u) CYTOPENIAS; NO OR RARE BLASTS; NO AUER RODS; UNILINEAGE DYSPLASIA; <5% BLASTS; NO AUER RODS 5q-SYNDROME ANEMIA; NORMAL/INCREASED PLATELET COUNT; <5%BLASTS NORMAL/INCREASED MEGAKARYOCYTES; <5%BLASTS; NO AUER RODS CHILDHOOD MDS < 2 % BLASTS DYSPLASTIC CHANGES IN >10 % NEUTROPHILS DYSPLASTIC CHANGES IN > 10 % ERYTHROID PRECURSORS DYSPLASTIC CHANGES IN > 10 % GRANULOCYTE PRECURSORS MICROMEGAKARYOCY TES,DYSPLASTIC CHANGES IN MGKS
- 19. DIFFERENCES BETWEEN WHO AND FAB The WHO system • Makes use of cytogenetic findings. • The category of RAEB-t was eliminated as it got included within AML(>20%blasts). • CMML was removed and put in a new category of myelodysplastic/myeloproliferative diseases. • Adds the subtypes 5q syndrome and unclassifiable MDS. • Recognizes the prognostic importance of % of bone marrow blasts
- 20. PATHOGENESIS MDS : a stem cell disorder • It represents manifestation of the malignant transformation of myeloid stem cell • The abnormal cells in MDS are clones derived from an abnormal stem cell Apoptosis in MDS • Mechanism appears to be one of increased apoptosis of haemopoietic precursors in the marrow, • Presence of cytopenias despite a typically hypercellular bone marrow. • For those patients undergoing leukaemic transformation,the cytopenias arise due to maturation block of the malignant cells • Apoptosis is more prominent in early MDS, such as RA and RARS, than in advanced MDS with excess myeloblasts
- 21. Ineffective Hematopoiesis • Colony forming capacities of pleuripotent stem cells and their progeny are low or absent • Lower level of GM-CSF, M-CSF,IL 6 .IL 3, • CFU- GM less responsive to both G-CSF & GM-CSF • More dramatic in pts with RAEB or RAEB –t Immunological abnormalities in MDS • Commonly encountered in MDS, suggesting that they may play a role in the aetiology and pathogenesis of the disease.
- 22. • Particularly apparent in cases of hypoplastic MDS that share a number of features in common with aplastic anaemia, notably clinical presentation with macrocytosis and varying levels of dyserythropoiesis • Acquired mutations in the PIG-A gene characteristic of paroxysmal nocturnal haemoglobinuria (PNH) are also encountered Angiogenesis • Autocrine production of angiogenic molecules promotes expansion of leukemic clone • Vascular endothelial growth factor(VEGF) and its receptor VEGFR-1 And VEGFR-2 is overexpressed
- 23. Molecular basis of MDS • MDS is a preleukaemic disorder characterized by impaired cellular differentiation that has the potential to transform to AML if this abnormality is coupled to enhanced survival and proliferation. • The common chromosomal abnormalities found in MDS include loss of Y, monosomy 5, monosomy 7,trisomy 8, 20q – , abnormalities of 11q23, and deletions of 17p, 12p, 13q and 11q among others.
- 25. Clinical features • Asymptomatic - Many patients are diagnosed on routine laboratory screening • Fatigue, weakness, angina - as a result of anemia. • Infections most commonly bacterial, predominate with skin being the most common site. This is the most common cause of mortality and morbidity in MDS.
- 26. • Autoimmune abnormalities (uncommon) - Seen in 14 % of the patients. Most common is cutaneous vasculitis. • Cutaneous manifestations of MDS Sweet syndrome Granulocytic sarcoma
- 27. LABORATORY FINDINGS • PERIPHERAL BLOOD PICTURE • ERYTHROCYTES Anemia-variable RBC’s are macrocytic, macro ovalocytes seen. Reticulopenia Elliptocytosis, tear drop cells, stomatocytes seen. Basophilic stippling,Howell-Jolly bodies, normoblasts.
- 28. • OTHER ERYTHROCYTE CHANGES • Increase in fetal hemoglobin • Altered A,B, antigens on the surface.
- 29. • LEUKOCYTES Neutropenia - 2nd most common cytopenia Dysgranulopoiesis is seen by agranular or hypogranular neutrophils Persistent basophilia of cytoplasm Hyposegmentation (pseudo Pelger-Huet) of the nucleus Hypersegmentation of the nucleus is seen sometimes
- 30. • OTHER DEFECTS Enzyme defects such as Decreased myeloperoxidase, Decreased leukocyte alkaline phosphatase . Causes functional impairment of the neutrophils like defective bactericidal, phagocytic and chemotactic properties.
- 31. PLATELETS • Varying degree of thrombocytopenia • Platelets may show agranular/hypogranular cytoplasm • Giant platelets are seen • Micromegakarocytes are seen. They have a single lobe nucleus with cytoplasmic tags. Nucleus shows densely clumped chromatin.
- 32. BONE MARROW ASPIRATE • Well stained BM aspirate smears • At least 500 cells are to be counted • At least 30 megakaryocyte to be evaluated • Dysplastic features should be present in > 10 % cells CELLULARITY In most cases it is hypercellular But is hypocellular in Hypoplastic MDS
- 33. Erythropoeisis • Usually megaloblastic erythropoeisis • Feature of dyserthropoiesis • Some precursors may show Howell Jolly bodies • Vacoulization , basophilia and poor hemoglobinisation • Ring sideroblasts • PAS stain – may show granular positivity of normoblasts • Advanced cases – erythroid hypoplasia seen
- 34. Granulopoiesis • Usually myeloid hyperplasia • Hypogranulrity and hyposegmentation • Maturation arrest in myelocyte stage may be seen • Abnormal staining of primary granules seen in myelocyte & promyelocytes. Granules may be larger than normal or completely absent. • Irregular cytoplasmic basophilia seen
- 35. • Thrombopoiesis • Usually normal or megakaryocytic hyperplasia • Micromegakaryocytes, multinucleated megakaryocytes , & hypolobated megakaryocytes • Presence of > 10 % Micromegakaryocytes in a population suggests MDS • CD 61 staining
- 36. TREPHINE BIOPSY IN MDS Useful for determining • Cellularity of marrow • Abnormal localization of immature precursors (ALIP) • Reticulin fibrosis, Megakaryocytic dysplasia, Lymphoid aggregate • Hypoplastic MDS • Increases the diagnostic accuracy & helps in refining the IPSS score
- 37. EVALUATION OF SUSPECTED MDS • HISTORY Prior exposure to CT/RT Recurrent infections, bleeding gums • EXAMINATION Pallor/ bruising Splenomegaly • BLOOD COUNTS Hb, TLC, platelet count reticulocyte count • BLOOD FILM Macrocytosis, cytopenia, neytrophilia, monocytosis pseudo pelger huet anomaly,hypogranular neutrophils
- 38. • BONE MARROW ASPIRATE • BONE MARROW TREPHINE BIOPSY • BONE MARROW CYTOGENETICS ANALYSIS • EXCLUSION OF REACTIVE CAUSES OF DYSPLASIA Megaloblastic anaemia HIV infection Recent cytotoxic therapy Alcoholism Recurrent intercurrent infection
- 39. MINIMAL DIAGNOSTIC CRITERIA FOR MDS IN CHILDREN • At least two of the following Sustained unexplained cytopenia( neutropenia, thrombocytopenia , anemia) At least bilineage morphological myelodysplasia Acquired clonal cytogenetic abmormality in hematopoietic cells Increased blasts > 5%
- 40. DIFFERENTIAL DIAGNOSIS 1. Vitamin B 12 and folic acid deficiency 2. AML M6 3. HIV infection 4. Parvo virus B 19 infection 5. Exposure to arsenic and other heavy metals 6. Congenital Dyserythropoietic anemia 7. Paroxysmal nocturnal hemoglobinemia 8. G- CSF Therapy
- 41. IPSS risk-based classification system Marrow blast percentage: < 5 0 5-10 0.5 11-20 1.5 21-30 2.0 Cytogentic features Good prognosis 0 (–Y, 5q- , 20q-) Intermediate prognosis 0.5 (+8, miscellaneous single abnormality, double abnormalities) Poor prognosis 1.0 (abnor. 7, complex- >3 abnor.) Cytopenias None or one type 0 2 or 3 type 0.5
- 42. INTERNATIONAL PROGNOSTIC SCORING SYSTEM(IPSS) RISK SCORE AML TRANSFOR MATION % MEDIAN SURVIVAL (YEARS) LOW 0 19 5.7 INTERMEDIATE -1 0.5- 1.0 30 3.5 INTERMEDIATE -2 1.5 -2.0 33 1.2 HIGH 2.5 45 0.4
- 44. Applying WHO to Indian settings • Indian MDS differs from MDS of West Younger age at presentation Cytopenias more severe at presentation Patients opted for less aggressive treatment Poorer treatment outcomes Infections, nutritional disorders: commoner Follow-up: Not always available Majority of pts had MDS-RA, MDS RAEB 1 & 2
- 45. Treatment strategy (lower risk MDS) ★Improve blood cytopenias ★ Improve quality of life ★ Delay disease progression ★ Prolong survival The therapeutic strategy remains largely based on the IPSS(R).
- 46. Treatment strategy (Higher risk MDS) ★Improve blood cytopenias ★ Improve quality of life ★ Delay disease progression ★ Prolong survival
- 47. Treatment of patients with MDS: goals and options Clinically significant cytopenia(s) Supportive care Goals ● To reduce morbidity/mortality due to cytopenias ● To improve QoL Active therapy Goals ● To alter the natural history of MDS ● To improve survival ● To improve QoL ● To alleviate complications Transfusions (+ iron chelation) Growth factors Treatment of infections HSCT Chemotherapy • Intensive • Low-dose Hypomethylating agents Azacitidine/decitabine Lenalidomide, immunosuppressive Rx HSCT = haemopoietic stem cell transplantation; QoL = quality of life.
- 48. CONCLUSION • MDS can be effectively diagnosed and classified as per WHO 2008 classification • MDS diagnosis and classification is currently in a transitional phase from reliance almost entirely on cell morphology supplemented by cytochemistry and G-banded karyotyping, towards a new era in which molecular and perhaps immunophenotypic findings will be fully incorporated • But in developing countries it is essential to rule out infections/ nutritional deficiencies especially among the elderly before considering MDS
- 49. 1 .Which ONE of these is the most common finding in myelodysplastic syndromes (MDS)? A Hypocellular bone marrow and reduced blood cell counts B Hypocellular bone marrow and increased blood cell counts C Hypercellular bone marrow and reduced blood cell counts D Hypercellular bone marrow and increased blood cell counts
- 50. 2.Which ONE of these is NOT a feature of peripheral blood cells in myelodysplastic syndromes? A Increased haemoglobin concentration in red cells B Macrocytic red cells C Hypogranular neutrophils D Bilobed nucleus in neutrophils
- 51. 3. What percentage of blast cells in the bone marrow would indicate a diagnosis of acute myeloid leukaemia rather than myelodysplasia? A 5 B 10 C 20 D 30
- 52. 4. Which ONE of these is NOT a diagnostic subtype of myelodysplasia? A Refractory anaemia with ring sideroblasts B Refractory anaemia with excess blasts C Refractory cytopenia with unilineage dysplasia D X‐linked sideroblastic anaemia
- 53. 5. What is the most likely treatment for a 75-year-old patient with refractory anaemia and haemoglobin 9 g/dL? A Azacytidine B Stem cell transplantation C Lenolidamide D Trial of erythropoietin
- 54. 6. Which ONE of these is the most powerful predictive factor in patients with myelodysplastic syndrome? A Serum β2‐microglobulin B Gender C The number of blasts in the bone marrow D The level of anaemia
- 55. 7.Which ONE of these is the most likely clinical picture in a patient with myelodysplastic syndrome associated with isolated del(5q)? A A man with Hb < 13 g/dL and a platelet count of 600 × 109/L B A woman with Hb <10 g/dL and platelet count of 600 × 109/L C A woman with Hb <13 g/dL and platelet count 100 × 109/L D A man with Hb <10 g/dL and platelet count 100 x 109/L
- 56. 8.In which patients with myelodysplastic syndrome is ciclosporin most likely to be effective? A Low risk disease with hypocellular bone marrow B Low risk disease with hypercellular bone marrow C High risk disease with hypocellular bone marrow D High risk disease with hypercellular bone marrow
- 57. 1.>15% ringed sideroblasts 2.>30% ringed sideroblasts 3.dyshematopoiesis in all three cell lineages 4.pancytopenia 9.In addition to the number of blasts, what other criterion is essential for a diagnosis of RARS?
- 58. A. lymphocytosis in the peripheral blood B.mature granulocytosis in the peripheral blood C.hypocellular bone marrow D. monocytopenia 9.CMML differs from the other subgroups of MDS because it has?
- 59. 1.macrocytic, normochromic 2.normocytic, normochromic 3.microcytic, hypochromic 4.normocytic, hypochromic 10.The type of anemia usually seen in MDS is?
- 60. 1.Leukopenia 2.Thrombocytopenia 3.Anemia 4.a combination of two of the above 11.The most common "cytopenia(s)" seen in MDS is/are?
- 61. 1.megaloblastoid development 2.impaired hemoglobinization 3.pseudo Pelger-Huet cells 4.agranular cytoplasm 12.The most common dyserythropoietic finding in the bone marrow in MDS
- 62. RA RARS RAEB RAEB-t USE THE FOLLOWING INFORMATION TO ANSWER QUESTIONS 12-14: A patient presents with the following laboratory data: RBC 2.30 x 1012/L HGB 78 g/L HCT 0.24 L/L MCV 104 fL RDW 20 WBC 8.5 x 109/L PLT 140 x 109/L The differential was normal except for 2% metamyelocytes. Oval macrocytes, a few abnormal NRBC, and a few siderocytes were seen. The bone marrow contained 3% blasts and exhibited hypercellularity with megaloblastoid development in erythroid cells.
- 63. 12.What is the most probable MDS subgroup? 1.RA 2.RARS 3.RAEB 4.RAEB-t
- 64. 1.aplastic anemia 2.megaloblastic anemia 3.iron-deficiency anemia 4.anemia of chronic disease 13.What other hematologic disorder does this peripheral blood picture resemble?
- 65. 1.serum lysozyme 2.serum ferritin 3.serum folate and vitamin B12 4.combined esterase stain 14.In reference to question #13, what laboratory test(s) would be helpful to distinguish the two disorders?
Editor's Notes
- 45
- 46
- 47