Myopathies
•Download as PPTX, PDF•
75 likes•16,634 views

this presentation briefly discus about muscle and its related disorder. some myopathies which are common are cover here in an approach to provide basis of the same disease and treatment. this ppt is basically from chapter 32 zakazewski.










































Myopathies
- 2. DEFINITION • Myopathy is a neuromuscular disorders in which the primary symptom is muscle weakness due to dysfunction of muscle fiber. • Hetrogenous disorder • Myopathies disorder affecting Channel Structure Metabolism of skeletal muscle
- 3. EPIDEMIOLOGY • Worldwide incidence of all inheritable myopathies is about 14% • Overall incidence of muscular dystrophy is about 63 per 1 million. • Worldwide incidence of inflammatory myopathies is about 5–10 per 100,000 people. • More common in women • Corticosteroid myopathy is the most common endocrine myopathy and endocrine disorders are more common in women • Overall incidence of metabolic myopathies is unknown.
- 6. MOTOR UNIT • A motor unit is made up of a motor neuron and all the muscle cells it stimulates. • vary in size • Small motor units for precise, small movements • large motor units for gross movements. • The number of cells within a motor unit determines the degree of movement when the motor unit is stimulated. • Muscle tone is maintained by asynchronous stimulation of random motor units.
- 7. CLASSIFICATION OF MUSCLE FIBER TYPE
- 8. CLINICAL EVALUATION History Muscle Weakness • Proximal Muscles>distal muscles • Fatigue • Difficulty rising from a chair, floor, tub • Difficulty with stairs • Difficulty with overhead tasks • Respiratory muscles • Bulbar weakness- speech, swallowing, oculomotor, facial Fatigue Muscle pain, cramp, stiffness Paresthesia and dysesthesias
- 9. PHYSICAL EXAMINATION • Demonstration and quantification of weakness • Muscle bulk • Muscle palpation • Fatigue • Muscle tone • Reflexes • Sensation
- 10. CLASSIFICATION OF MYOPATHY • Inherited • Acquired
- 11. MUSCULAR DYSTROPHY • Muscular dystrophy refers to a group of hereditary progressive diseases each with unique phenotypic and genetic features. • Dystrophinopathies: Duchenne’s Muscular Dystrophy and Becker’s Muscular Dystrophy • Facioscapulohumeral muscular dystrophy • Emery-Dreifus muscular dystrophy • Limb-girdle muscular dystrophy • Myotonic dystrophy
- 12. DUCHENNE’S MUSCULAR DYSTROPHY • Duchenne’s muscular dystrophy (DMD) also called pseudo hypertrophic muscular dystrophy. • It is an X linked recessive disorder. • Dystrophin is deficient. • The incidence of DMD is 1 in 3500 male births worldwide, • DMD clinical feature: It is present at birth but becomes evident at 3-5 years. Gower’s maneuver Joint contractures, scoliosis, decreased pulmonary functions. By 16 to 18 years patients die of severe pulmonary infections or aspiration pneumonia. Respitatory failure in 2nd or 3rd decade. IQ <~1 SD of the mean.
- 13. GOWER’S MANEUVER
- 15. BECKER’S MUSCULAR DYSTROPHY • X linked recessive inheritance. • Less severe form. • Dystrophin muscle protein is deficient. • The incidence of BMD is 5 in 100,000. Clinical features: • Muscle wasting resembles Duchenne’s. • Proximal muscle weakness of lower extremities occur first. • Onset 5-15 years or even 3rd to 4th decade. • Patients may survive till 4th or 5th decade. Laboratory findings are similar to that of Duchenne’s muscular dystrophy.
- 18. FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY • Third most common after DMD and myotonic • Onset of occurrence is childhood or young adulthood (age 3 to 44) • Prevelence ranging from 1 in 20,000 to 1 in 455,000 • Autosomal dominant linked to chromosome 4q35 Clinical feature • Onset is incidious • Difficulty in overhead activity • Humeral muscle affected with sparing of FA muscle give ‘popeye’ appereance • Positive beevor sign
- 20. EMERY-DREIFUSS MUSCULAR DYSTROPHY • Caused by mutation in STA gene which code for key nuclear protein or LMNA gene which code for lamins A and C. • The X-linked form affects males, but females may present with isolated cardiomyopathy and therefore require cardiac evaluation. • EDMD is characterized by a triad of clinical features: (1) early contractures in the elbows, Achilles tendon, and posterior cervical spinal muscles (2) slowly progressive muscle weakness, which begins in a humeroperoneal distribution; (3) cardiac abnormalities such as cardiomyopathy and conduction defects.
- 22. LIMB-GIRDLE MUSCULAR DYSTROPHY • Genetically heterogeneous group of disorders with an autosomal dominant (LGMD 1) or autosomal recessive (LGMD 2) mode of inheritance. • The prevalence is approximately 8.1 in 1 million inhabitants. • Underlying pathophysiology is unknown. Clincal features; • Lower limb and pelvic girdle weakness, later UL weakness and scapular winging • Facial and extraocular muscle spared • Diaphragmatic weakness • Cardiac abnormality
- 24. MYOTONIC DYSTROPHY • Autosomal dominant mode of inheritance mapped to chromosome 19q13,53 which codes for the myotonic dystrophy protein kinase (DMPK). • Incidence of myotonic dystrophy is approximately 13.5 in 100,000 live births, and the prevalence is 3 to 5 per 100,000.14. Clinical features: • Slow progressive weakness of face, jaw and distal limb • Frontal baldness, ptosis, and atrophy in the temporalis and masseter muscles result in a characteristic “hatchet-faced” appearance. • Dysarthria and dysphagia • Myotonia • Warm-up phenomenon
- 27. CONGENITAL MYOPATIES • Heterogeneous group of nonprogressive or slowly progressive muscle disorders that usually present in the neonatal period. • Not considered muscular dystrophies • present with generalized weakness, hyporeflexia and hypotonia •Delayed motor milestone and decreased muscle bulk •A recent review article (North K., 2008) divided the congenital myopathies based on genetic and morphological features into 4 main groups: • Affected both sexes equally • laboratory finding: normal or minimally elevated CK level, emg finding can show polyphasic motor unit potential • Management is multidisciplinary apporach Myopathies with protein accumulation Nemaline myopathy Myosin storage myopathy Cap disease Reducing body myopathy Myopathies with cores Central core disease Core-rod myopathy Multiminicore disease Myopathies with central nuclei Myotubular myopathy Centronuclear myopathy Myopathies with fiber size variation Congenital fiber type disproportion
- 29. CENTRAL CORE MYOPATHY • Autosomal dominant • Mutation in ryanodine receptor gene (RYR1) • Higher risk for malignant hyperthermia • Presentation: hypotonia, decreased muscle bulk, slender frame, and symmetrical weakness. • Weakness can varies and predominantly affect proximal muscle of lower limb • Motor milrstone delayed but able to walk by age 3 to 4. • No CNS abnormality • Muscle biopsy shows characteristic structural alterations within the center of type 1 muscle fibers known as cores. These cores are single, centrally located, and circular.
- 30. NEMALINE MYOPATHY • Nema in Greek means thread. • An autosomal dominant, recessive, or sporadic mode of inheritance • Caused by mutations in genes that code for proteins that are responsible for the development and function of the Z-disks, including actin, troponin, nebulin, and tropomyosin. • The disease may present as three phenotypes and the most severe phenotype is the infantile onset, common phenotype is childhood onset. • Neonates present with hypotonia, feeding and respiratory difficulty, children can have delayed milestones • The long, narrow facies, higharched palate, and openmouthed appearance due to a prognathous jaw. Pectus excavatum, kyphoscoliosis, pes cavus, and clubfoot deformities.
- 31. • Muscle biopsy reveals characteristic threadlike structures (rods) that consist of Z-disk protein material • No specific pharmacological treatment is available. Rehabilitation should focus on maintaining function and preventing deconditioning through mild exercise and physical therapy.
- 32. MYOTUBULAR/ CENTRONUCLEAR MYOPATHY • The disease can be X-linked, mapped to chromosome Xq28 at a locus coding for myotubularin. • It can have an autosomal dominant or recessive mode of inheritance Three variants: • A neonatal form presents with severe hypotonia and weakness at birth. • The late infancy–early childhood form presents with delayed motor milestones. • Later, difficulty with running an stair climbing • A marfanoid, slender body habitus, long narrow face, and high-arched palate are typical. • Presentation at birth: hypotonia, feeding difficulty, respiratory distress, bilateral ptosis, limited eye movements, and absent tendon reflexes. • Early childhood: Gait is waddling and hyperlordotic • Facial dysmorphic features are often present • Muscle biopsy shows myonuclei in the center of the muscle fibers • Treatment: multidisciplinary approach
- 33. Abundance of centrally located nuclei including majority muscle fibre (mostly in type 1 fibres)
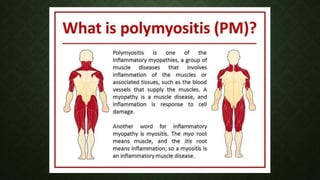
- 34. INFLAMMATORY MYOPATHIES: POLYMYOSITIS AND DERMATOMYOSITIS • Idiopathic inflammatory disorders. • Usually presents in those older than age 20. • Female-to-male ratio is approximately 2:1. • Pathogenesis:
- 36. CLINICAL PRESENTATION • Present with a progressive, symmetrical, proximal (i.e., more than distal) pattern of muscle weakness. • Muscle pain and tenderness • Later neck, swallowing, and respiratory muscles may become affected • Arthralgia (50% patient) • DM present with erythmatous skin lesion, Heliotrope rash, Gottron’s rash is a violaceous, raised, scaly rash over the knuckles. • PM and DM are associated with abnormalities in the cardiac and pulmonary systems. • Patients with PM or DM may have a connective tissue disease, such as rheumatoid arthritis, systemic lupus erythematous, scleroderma, or Sjögren’s syndrome.
- 38. METABOLIC MYOPATHIES • Heterogeneous group of disorders caused by genetic defects that compromise muscle energy production. • Enzyme dysfunction can result in an inadequate supply of ATP. • At least 14 enzyme defects that affect glycogen synthesis, glycogenolysis, and glycolysis have been described. • Other metabolic myopathies affect lipid metabolism. • the more common metabolic disorders that result in myopathies, including myophosphorylase deficiency, phosphofructokinase (PFK) deficiency, debrancher enzyme deficiency, and acid maltase deficiency
- 40. ENDOCRINE MYOPATHIES • Frequently manifest with muscular impairment. • The features of endocrine myopathies most amenable to rehabilitation intervention include muscle weakness and atrophy. • Exercises, orthoses, or assistive devices may be necessary, depending on the severity of the deficits.
- 41. Endocrine myopathy Features Clinical presentation Diagnosis Treatment Steroid myopathy •Most common •incidence is 2.4% to 21% •Women at risk •Insidious onset •proximal muscle weakness and atrophy •greater involvement of the lower limbs •Normal CK. •Muscle biopsy shows atrophy of type II fibers •Stoping and reduce the dose •Strenth training to overcome weakness Hyperthyroidism •82% affected •Female more than male •Pathogenesis: enhanced muscle protein catabolism with muscle amino acid by the elevated thyroxine •Weakness •Muscle atrophy •Fatigue, myalgia, and exercise intolerance. •Respiratory muscle involvement •Dysphagia and dysphonia. •Tendon reflexes:normal or brisk. •elevated T3 and T4 and a low TSH •Needle EMG is usually normal, fasciculations may be present. •Active exercises Hypothyroidism •Proximal muscle weakness, stiffness, fatigue, and slowed movements •myoedema •CK usually is elevated •T3 and T4 are depressed, TSH is elevated. •Treatment of the underlying thyroid dysfunction
- 42. TOXIC MYOPATHIES
- 43. REFERENCES: • Myopathy: SUJATA MAHARATHI, DEMONSTRATOR, PHYSIOTHERAPY • Harrison's Principle of Internal Medicine 19th Edition • Zackizweiski Musculoskeletal rehabilitation; chapter 32: Muscle Disease and Dysfunction; SABRINA PAGANONI, ANNE-MARIE THOMAS, WALTER R. FRONTERA 2nd edition.
Editor's Notes
- Pathogenesis An autoimmune etiology of the inflammatory myopathies is indirectly supported by an association with other autoimmune or connective tissue diseases; the presence of various autoantibodies; an association with specific major histocompatibility complex (MHC) genes; demonstration of T cell-mediated myocytotoxicity or complement-mediated microangiopathy; and a response to immunotherapy. Autoantibodies and Immunogenetics Various autoantibodies against nuclear antigens (antinu-clear antibodies) and cytoplasmic antigens are found in up to 20% of patients with inflammatory myopathies. The antibodies to cytoplasmic antigens are directed against ribonucleoproteins involved in protein synthesis (anti-synthetases) or translational transport (anti-signal- recognition particles).The antibody directed against the histidyl-transfer RNA synthetase, called anti-Jo-1, accounts for 75% of all the anti-synthetases and is clinically useful because up to 80% of patients with anti-Jo-1 antibodies have interstitial lung disease. Some patients with the anti-Jo-1 antibody also have Raynaud’s phenomenon, nonerosive arthritis, and the MHC molecules DR3 and DRw52. DR3 haplotypes (molecular designation DRB1*0301,DQB1*0201) occur in up to 75% of patients with PM and IBM, whereas in juvenile DM there is an increased frequency of DQA1*0501 (Chap. 2). Immunopathologic Mechanisms In DM, humoral immune mechanisms are implicated, resulting in a microangiopathy and muscle ischemia (Fig. 16-1). Endomysial inflammatory infiltrates are composed of B cells located in proximity to CD4 T cells, dendritic cells, and macrophages; there is a relative absence of lymphocytic invasion of nonnecrotic muscle fibers. FIGURE 16-1 Immunopathogenesis of dermatomyositis. Activation of complement, possibly by autoantibodies (Y), against endothelial cells and formation of C3 via the classic or alternative pathway. Activated C3 leads to formation of C3b, C3bNEO, and membrane attack complexes (MAC), which are deposited in and around the endothelial cell wall of the endomysial capillaries. Deposition of MAC leads to destruction of capillaries, ischemia, or microinfarcts most prominent in the periphery of the fascicles, and perifascicular atrophy. B cells, CD4 T cells, and macrophages traffic from the circulation to the muscle. Endothelial expression of vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule (ICAM) is induced by cytokines released by the mononuclear cells. Integrins, specifically very late activation antigen (VLA)-4 and leukocyte function-associated antigen (LFA)-1, bind VCAM and ICAM and promote T-cell and macrophage infiltration of muscle through the endothelial cell wall. Activation of the complement C5b-9 membranolytic attack complex is thought to be a critical early event that triggers release of proinflammatory cytokines and chemokines, induces expression of vascular cell adhesion molecule (VCAM) 1 and intracellular adhesion molecule (ICAM) 1 on endothelial cells, and facilitates migration of activated lymphoid cells to the perimysial and endomysial spaces. Necrosis of the endothelial cells, reduced numbers of endomysial capillaries, ischemia, and muscle-fiber destruction resembling microinfarcts occur. The remaining capillaries often have dilated lumens in response to the ischemic process. Larger intramuscular blood vessels may also be affected in the same pattern. Residual perifascicular atrophy reflects the endofascicular hypoperfusion that is prominent in the periphery of the muscle fascicles. By contrast, in PM and IBM a mechanism of T cell-mediated cytotoxicity is likely. CD8 T cells, along with macrophages, initially surround and eventually invade and destroy healthy, nonnecrotic muscle fibers that aberrantly express class I MHC molecules. MHC-I expression, absent from the sarcolemma of normal muscle fibers, is probably induced by cytokines secreted by activated T cells and macrophages. The CD8/MHC-I complex is characteristic of PM and IBM; its detection can aid in confirming the histologic diagnosis of PM, as discussed below. The cytotoxic CD8 T cells contain perforin and granzyme granules directed towards the surface of the muscle fibers and capable of inducing myonecrosis. Analysis of T-cell receptor molecules expressed by the infiltrating CD8 cells have revealed clonal expansion and conserved sequences in the antigen-binding region, both suggesting an antigen-driven T-cell response. Whether the putative antigens are endogenous (e.g., muscle) or exogenous (e.g., viral) sequences is unknown. Viruses have not been identified within the muscle fibers. Co-stimulatory molecules and their counterreceptors, which are fundamental for T-cell activation and antigen recognition, are strongly upregu-lated in PM and IBM. Key molecules involved in T cell-mediated cytotoxicity are depicted in Fig. 16-2. FIGURE 16-2 Cell-mediated mechanisms of muscle damage in polymyositis (PM) and inclusion body myositis (IBM). Antigen-specific CD8 cells are expanded in he periphery, cross the endothelial barrier, and bind directly to muscle fibers via T-cell receptor (TCR) molecules that recognize aberrantly expressed MHC-I. Engagement of co-stimulatory molecules (BB1 and ICOSL) with their ligands (CD28, CTLA-4, and ICOS) along with ICAM-1/LFA-1, stabilize the CD8-muscle fiber interaction. Metalloproteinases (MMP) facilitate the migration of T cells and their attachment to the muscle surface. Muscle fiber necrosis occurs via perforin granules released by the autoaggressive T cells. A direct myocytotoxic effect exerted by the cytokines interferon (IFN) γ, interleukin (IL) 1, or tumor necrosis factor (TNF) α may also play a role. Death of the muscle fiber is mediated by necrosis. MHC class I molecules consist of a heavy chain and a light chain [ß2 microglobulin (ß2m)] complexed with an antigenic peptide that is transported into the endoplasmic reticulum by TAP proteins.