Este documento resume los conceptos clave de farmacocinética y su aplicación durante el embarazo. Explica los procesos de absorción, distribución, metabolismo y excreción de los fármacos y cómo estos procesos pueden verse afectados por los cambios fisiológicos durante la gestación. Se describen factores como el pH gástrico, la motilidad gastrointestinal, el volumen sanguíneo y los niveles de proteínas plasmáticas, que influyen en la biodisponibilidad y eliminación de los medicamentos. El
0 calificaciones0% encontró este documento útil (0 votos)
8 vistas33 páginas
Este documento resume los conceptos clave de farmacocinética y su aplicación durante el embarazo. Explica los procesos de absorción, distribución, metabolismo y excreción de los fármacos y cómo estos procesos pueden verse afectados por los cambios fisiológicos durante la gestación. Se describen factores como el pH gástrico, la motilidad gastrointestinal, el volumen sanguíneo y los niveles de proteínas plasmáticas, que influyen en la biodisponibilidad y eliminación de los medicamentos. El
Este documento resume los conceptos clave de farmacocinética y su aplicación durante el embarazo. Explica los procesos de absorción, distribución, metabolismo y excreción de los fármacos y cómo estos procesos pueden verse afectados por los cambios fisiológicos durante la gestación. Se describen factores como el pH gástrico, la motilidad gastrointestinal, el volumen sanguíneo y los niveles de proteínas plasmáticas, que influyen en la biodisponibilidad y eliminación de los medicamentos. El
Este documento resume los conceptos clave de farmacocinética y su aplicación durante el embarazo. Explica los procesos de absorción, distribución, metabolismo y excreción de los fármacos y cómo estos procesos pueden verse afectados por los cambios fisiológicos durante la gestación. Se describen factores como el pH gástrico, la motilidad gastrointestinal, el volumen sanguíneo y los niveles de proteínas plasmáticas, que influyen en la biodisponibilidad y eliminación de los medicamentos. El
Descargue como PPTX, PDF, TXT o lea en línea desde Scribd
Descargar como pptx, pdf o txt
Está en la página 1/ 33
UNIVERSIDAD NACIONAL MAYOR DE SAN MARCOS
(Universidad del Perú, DECANA DE AMERICA)
FACULTAD DE MEDICINA ESCUELA ACADÉMICO PROFESIONAL DE OBSTETRICIA
Farmacología General y Especializada
(16 de agosto de 2023)
Farmacocinética y Gestación
Dr. Hugo Justil Guerrero
Administración vía oral
¿Habrá diferencia en la eliminación de los fármacos en
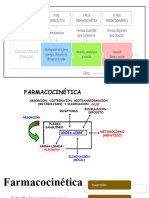
cada persona? CONTENIDO 1. PASO DE FÁRMACOS A TRAVÉS DE LAS MEMBRANAS BIOLÓGICAS.
2. INTERRERACIÓN DE LA ABSORCIÓN, DISTRIBUCIÓN, UNIÓN,
METABOLISMO Y EXCRECIÓN DE UN FÁRMACO.
3. LIBERACIÓN.
4. ABSORCIÓN BIODISPONIBILIDAD VÍAS DE ADMINISTRACIÓN.
5. DISTRIBUCIÓN
6. METABOLISMO
7. EXCRESIÓN PASO DE FÁRMACOS A TRAVÉS DE LAS MEMBRANAS BIOLÓGICAS.
• Transporte pasivo • Transporte activo. Gasto de energía
Goodman & Gilman 13ª Edición
1. Difusión pasiva Acido débil Aspirina pka ≈ 3,5
pKa ≈ 3,5
[] 1) pH del medio. 2) pKa del Fármaco. 2. Difusión facilitada La fuerza que impulsadora está mediado por el gradiente electroquímico del soluto transportado.
Rodrigo-Ayala et al (2020). https://doi.org/10.24245/gom. v88i3.3598
3. Transporte activo Requieren de energía. Capacidad de transporte contra gradiente electroquímico, saturación.
Astudillo-de la Vega et al.(2010). Papel de la quimioresistencia en
tumores sólidos INTEREACCIÓN DE ABSORCIÓN, DISTRIBUCIÓN, METABOLISMO Y EXCRECIÓN DE UN FÁRMACO
Goodman & Gilman 13ª Edición
LIBERACIÓN
Fases:
Desintegración: Paso de formas sólidas a
partículas más pequeñas
Disolución: Paso de formas sólidas a solución.
Difusión: Paso del fármaco disuelto a través del
fluido. ABSORCIÓN
Paso del fármaco desde su lugar de administración al torrente
sanguíneo.
La absorción depende de:
1. Propiedades fisicoquímicas del fármaco.
2. Vía de administración 3. Forma y presentación del medicamento. ABSORCIÓN
Forma farmacéutica Sólido, líquido, gaseoso, semisólido, geles Anillo vaginal Tabletas Presentación Tabletas, grajeas, supositorios, jarabes, inhaladores, tabletas de liberación retardada, enemas, colutorios, cápsulas, etc.
Parches Inhaladores
Ungüento Cambios fisiológicos y gestación
Variable alterada Proceso farmacocinético Efecto
alterado
Disminución pH salival Absorción Alteración absorción de fármacos sublinguales
Hipoacidez estomacal Absorción Alteración de absorción de fármacos orales
Disminución de motilidad gastrointestinal Absorción Alteración de absorción de fármacos orales
Incremento de gasto cardiaco y flujo sanguíneo Absorción Incremento de biodisponibilidad del fármaco.
Incremento de hiperventilación Absorción Mayor velocidad de transporte de fármaco por
membrana alveolar BIODISPONIBILIDAD Fármacos: Colchicina Biodisponibilidad 45% Fracción de fármaco de forma activa llega a Fenitoína 70% la circulación y alcanza su lugar de acción Tetraciclina 80% (diana terapéutica).
¿Qué factores habrán influido en la biodisponibilidad?
5. Periodo de Latencia = Tiempo en que demora en llegar el fármaco a
5 la CME
6. AUC = Área bajo la curva. Representa la biodisponibilidad del
6 fármaco. DISTRIBUCIÓN
VOLUMEN DE DISTRIBUCIÓN (Vd)
Vd =
Relaciona la cantidad de fármaco (Q) y la concentración plasmática
del fármaco (Cp)
Hígado, riñón, cerebro y otros órganos perfundidos reciben la mayor
parte del fármaco.
Unión a proteínas plasmáticas: Albúmina y glucoproteína ácida alfa1.
Ejemplos MODELOS COMPARTIMENTALES DISTRIBUCIÓN DE FÁRMACOS EN LA UNIDAD MATERNO-PLACENTARIO-FETO CAMBIOS FISIOLÓGICOS Y GESTACIÓN Variable alterada Proceso farmacocinético Efecto alterado
Incremento de la perfusión Distribución Mayor llega a placenta
Incremento de agua corporal y liquido extracelular Distribución Menor concentración plasmática
Disminución de proteínas plasmáticas Distribución Fármaco libre en el plasma.
METABOLISMO El metabolismo de los fármacos tiene lugar en dos etapas:
Las reacciones de funcionalización que
tienen lugar en la fase I (pérdida de actividad farmacológica) Estas son: oxidación, reducción, hidrólisis y descarboxilación.
Las reacciones biosíntesis o conjugación
tienen lugar en fase II (lo hacen más polar al fármaco) Enlace covalente con acido glucorónido, sulfato de glutatión, aminoácidos o acetatos. CYP SUB FAMILIAS EN EL HÍGADO ALGUNOS FÁRMACOS METABILIZADOS VÍA CYP EXCRECIÓN Eliminación de un fármaco y sus metabolitos del organismo
2. No renal. a) Biliar. Principalmente lipofílicos, transporte activo, posibilidad de circulación enteropática (hígado – bilis – intestino – heces). b) Glándulas mamarias. Difusión pasiva, fármacos liposolubles, básicos (Cloranfenicol, tetraciclinas, fenitoína, Sulfamidas). c) Saliva. Depende del pka, solubilidad y fracción libre del fármaco. FACTORES QUE MODIFICAN LA EXCRECIÓN RENAL
Insuficiencia renal Disminución de la perfusión renal Edad Competencia con el transportador Modificación del pH de la orina. Cl = Ve/Cp ACLARAMIENTO (CL) O DEPURACIÓN Cl = Vd . Ke
• Habilidad para eliminar fármaco del sistema vascular a través de
• Alteración renal o hepática podrá disminuir el aclaramiento.
• Permite determinar la concentración en estado estable (Css) y
determinar la velocidad de administración.
• Dosis de fármaco de acuerdo al Cl plasmático.
VELOCIDAD DE ELIMINACIÓN Cinética orden “0”
20% 200 000
Velocidad de eliminación (mg/h)
30% 30 000
40% 4 000
500 Orden 1
50% 50
10 100 1000 10 000 100 000
6 7 8 10 10 10
Concentración (mg) TIEMPO DE VIDA MEDIA (t1/2) Tiempo en que la concentración plasmática de un Fármaco se reduce a la mitad. Determina el tiempo que tarda un Fármaco en eliminar (4 t1/2) Determina el tiempo que tarda en alcanzar estacionario (4 t1/2) ELIMINACIÓN ES PROCESO DE PRIMER ORDEN. DONDE EL TIEMPO DE ELIMINACIÓN ES APROXIMADAMENTE 4 T1/2 Eliminación de una dosis
100
50
25
12,5
6,25 t1/2 1 2 3 4 (horas) DOSIFICACIÓN
Establecimiento de la dosis y de los intervalos de dosificación
El objetivo es alcanzar la Concentración plasmática en el estado de equilibrio en el rango terapéutico DOSIFICACIÓN
Para una dosis:
Dosis = Cp (objetivo) x Vd
Para dosis múltiples se debe conocer:
Concentración mínima eficaz
Parámetros farmacocinéticos del medicamento: aclaramiento (Cl) y volumen de
distribución (Vd). ESTADO DE EQUILIBRIO O ESTADO ESTABLE (CSS) Inicial y autolimitado
194 197 200 187,5 Aproximadamente de 4 T1/2. 175 150 150 Permite interpretar la relación de la concentración y el efecto 100 94 97 98 87,5 terapéutico 75 50 50
1 2 3 4 5 7 CAMBIOS FISIOLÓGICOS Y GESTACIÓN Variable alterada Proceso farmacocinético Efecto alterado
Incremento de progesterona Metabolismo Inductor enzimático
Incremento de flujo renal y filtración glomerular Eliminación Incremento de aclaramiento y disminución de
concentración plasmática. pH orina básico Eliminación Incremento de excreción de los fármacos ácidos.