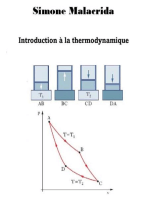
Chapitre 3-1
Chapitre 3-1
Télécharger au format pdf ou txt
Vous aimerez peut-être aussi
- CHAPITRE.2.Premier Principe de La ThermodynamiqueDocument12 pagesCHAPITRE.2.Premier Principe de La ThermodynamiquenajibPas encore d'évaluation
- ÉTAT THERMODYNAMIQUE - PDF PugerDocument13 pagesÉTAT THERMODYNAMIQUE - PDF Pugergeorges.kevin.henderson.681068732Pas encore d'évaluation
- Chapitre 2 PREMIER PRINCIPE DE LA THERMODYNAMIQUEDocument7 pagesChapitre 2 PREMIER PRINCIPE DE LA THERMODYNAMIQUEslimanePas encore d'évaluation
- Poly 2 WebDocument11 pagesPoly 2 WebMohammad ElbaghatiPas encore d'évaluation
- ST2 Thermodynamique GourariDocument38 pagesST2 Thermodynamique GouraritthPas encore d'évaluation
- Annexe Différentielle LogarithmiqueDocument14 pagesAnnexe Différentielle LogarithmiqueYounes EL BAHRAOUIPas encore d'évaluation
- Support de Cours Thermodynamique 2020Document59 pagesSupport de Cours Thermodynamique 2020ahlemm100% (1)
- Cours Et Exercices - ÉTAT THERMODYNAMIQUE PDFDocument8 pagesCours Et Exercices - ÉTAT THERMODYNAMIQUE PDFOussama El BouadiPas encore d'évaluation
- Premier Principe de La ThermodynamiqueDocument10 pagesPremier Principe de La ThermodynamiqueNardjes Ben100% (1)
- Chapitre 1 ThemodynamiqueDocument9 pagesChapitre 1 Themodynamiqueخالد ابن الوليدPas encore d'évaluation
- Chapitre 2 ThermodynamiqueDocument4 pagesChapitre 2 ThermodynamiqueWael MaatougPas encore d'évaluation
- 1er PrincipeDocument3 pages1er Principegamepas3moiPas encore d'évaluation
- Chapitre 3 Thermo NIBOUDocument11 pagesChapitre 3 Thermo NIBOUILHAM M'HARZIPas encore d'évaluation
- Notes de Cours 1er Principe ThermodynamiqueDocument7 pagesNotes de Cours 1er Principe Thermodynamiquehadil.souafPas encore d'évaluation
- Cours 3 PDFDocument7 pagesCours 3 PDFqhfrt5ckq7Pas encore d'évaluation
- Thermo SytemeDocument10 pagesThermo SytemeSparyoS 78Pas encore d'évaluation
- Chapitre-II, Le Premier Principe de La ThermodynamiqueDocument12 pagesChapitre-II, Le Premier Principe de La ThermodynamiqueSalima SouhilaPas encore d'évaluation
- Cours Thermodynamique - Chap 2Document36 pagesCours Thermodynamique - Chap 2Lavd LoghPas encore d'évaluation
- Rappels Premier PrincipeDocument42 pagesRappels Premier Principemohamedelallaouy86Pas encore d'évaluation
- Chapitre 0 - Quelques Réflexions IntroductivesDocument53 pagesChapitre 0 - Quelques Réflexions IntroductivesNassira F-hPas encore d'évaluation
- Chapitr03 PDFDocument9 pagesChapitr03 PDFZach LeitchPas encore d'évaluation
- Chapitre 2Document18 pagesChapitre 2Noel SomdaPas encore d'évaluation
- Les Fonctions D'étatDocument14 pagesLes Fonctions D'étatmathys.piarronPas encore d'évaluation
- Chapitre 4 Partie (1) Travail Et ChaleurDocument32 pagesChapitre 4 Partie (1) Travail Et ChaleurAhmed amine El MoustajirPas encore d'évaluation
- Thermodynamique 1Document16 pagesThermodynamique 1Soumaila SdgPas encore d'évaluation
- Chapitre II Le Premier Principe de La ThermodynamiqueDocument13 pagesChapitre II Le Premier Principe de La ThermodynamiqueImane MatiPas encore d'évaluation
- Chapitre III - Chimie II - ThermochimieDocument12 pagesChapitre III - Chimie II - Thermochimiekim namjoonPas encore d'évaluation
- Chap II Thermo (20-21) S1Document23 pagesChap II Thermo (20-21) S1medmm200430Pas encore d'évaluation
- Cours ThermoDocument30 pagesCours ThermoayadiPas encore d'évaluation
- Maghat Cours SVT Thermo Equilibre PDFDocument28 pagesMaghat Cours SVT Thermo Equilibre PDFfaslaPas encore d'évaluation
- Chap4 1er - PrincipeDocument7 pagesChap4 1er - Principeyouness.rqiqqPas encore d'évaluation
- Chapitre 3Document19 pagesChapitre 3ahmedsaidka5Pas encore d'évaluation
- Thermo chp2Document8 pagesThermo chp2Achraf BlankiPas encore d'évaluation
- Thermochimie2021 22Document12 pagesThermochimie2021 22sanogoalima715Pas encore d'évaluation
- Aero Chimie-Principe Conservation EnergieDocument7 pagesAero Chimie-Principe Conservation EnergieAouini IbtihelPas encore d'évaluation
- Chapitre III Second Principe de La ThermodynamiqueDocument18 pagesChapitre III Second Principe de La ThermodynamiqueImane MatiPas encore d'évaluation
- Chap3 Thermo1 2007Document42 pagesChap3 Thermo1 2007Dana CapbunPas encore d'évaluation
- Thermodynamique CH 2Document21 pagesThermodynamique CH 2jouaitiPas encore d'évaluation
- Cours Thermodynamique - Chap 3Document21 pagesCours Thermodynamique - Chap 3Lavd LoghPas encore d'évaluation
- Chapitre I - Rappels - Des - Notions - Fondamentales - Final - KhoubaDocument17 pagesChapitre I - Rappels - Des - Notions - Fondamentales - Final - Khoubabouchra boudjPas encore d'évaluation
- Thermodynamique NPDocument11 pagesThermodynamique NPMaroc EcoloadPas encore d'évaluation
- 3-Le Second Principe de La ThermodynamiqueDocument12 pages3-Le Second Principe de La ThermodynamiqueAntes de Partir, A.C.Pas encore d'évaluation
- Le Resume de Cours ThermodynamiqueDocument24 pagesLe Resume de Cours ThermodynamiqueYassine EL DahmiPas encore d'évaluation
- THERMOCHIMIEDocument21 pagesTHERMOCHIMIEdoulaminegueyePas encore d'évaluation
- 3-Premier Principe - 2Document17 pages3-Premier Principe - 2ik0302303Pas encore d'évaluation
- Cours Chimie 1ère Année Thermodynamiq 1 2023 2024Document11 pagesCours Chimie 1ère Année Thermodynamiq 1 2023 2024Salima OUADFELPas encore d'évaluation
- COURS 1 Thermo Medecine 2016Document10 pagesCOURS 1 Thermo Medecine 2016Anonymous 7InwoqPas encore d'évaluation
- Thermodynamique1 PDFDocument19 pagesThermodynamique1 PDFabdelmoumen.doudoPas encore d'évaluation
- Chapitre 5 Premier PrincipeDocument19 pagesChapitre 5 Premier PrincipeAhmed amine El MoustajirPas encore d'évaluation
- Thermodynamique PDFDocument4 pagesThermodynamique PDFRedouane AmianPas encore d'évaluation
- E3A Physique Et Chimie MP 2006 CorrigeDocument4 pagesE3A Physique Et Chimie MP 2006 CorrigenazirawilphrisnaPas encore d'évaluation
- Chapitre 2Document21 pagesChapitre 2Mohammed AmliehPas encore d'évaluation
- Rappel ThermoDocument7 pagesRappel ThermoBadis ZeghbaPas encore d'évaluation
- Rappel Thermo.Document7 pagesRappel Thermo.Meriem BerrachediPas encore d'évaluation
- Thermo Dy Nami QueDocument3 pagesThermo Dy Nami QueiiMouadPas encore d'évaluation
- La Thermodynamique PersoDocument26 pagesLa Thermodynamique Persojean-michel HuetPas encore d'évaluation
- 3 Premier PrincipeDocument21 pages3 Premier Principeyoulmafia99Pas encore d'évaluation
- Th. Cl. Chapitre VIDocument13 pagesTh. Cl. Chapitre VIAissiou NabilaPas encore d'évaluation
- A. Applications Des Principes de La Thermodynamique: A. I. Étude Des Systèmes FermésDocument14 pagesA. Applications Des Principes de La Thermodynamique: A. I. Étude Des Systèmes FermésMihnea GamanPas encore d'évaluation
- Exercices PC TC International 6 2Document13 pagesExercices PC TC International 6 2kamalPas encore d'évaluation
- Mon Bao en Physique 1ière C Et D - Armel Zambou KenfackDocument469 pagesMon Bao en Physique 1ière C Et D - Armel Zambou KenfackArmel ZAMBOU KENFACKPas encore d'évaluation
- Caractérisation de L'enrochement Du Barrage Romaine-2: MémoireDocument177 pagesCaractérisation de L'enrochement Du Barrage Romaine-2: MémoireKarim iderPas encore d'évaluation
- Partie I-Matériaux DiélectriquesDocument42 pagesPartie I-Matériaux DiélectriquesFares Sadi100% (3)
- ANGLES: ActivitésDocument6 pagesANGLES: ActivitésLs NegocesPas encore d'évaluation
- Devoir de Synthèse N°1 2ème Semestre - Math - Bac Informatique (2018-2019) MR Hatem BouadilaDocument3 pagesDevoir de Synthèse N°1 2ème Semestre - Math - Bac Informatique (2018-2019) MR Hatem Bouadilahassinadib0Pas encore d'évaluation
- Fondamentaux FichesEnseignants 548Document7 pagesFondamentaux FichesEnseignants 548eakgunduzbalcikPas encore d'évaluation
- Isolation Thermique Hygrométrie HTMDocument6 pagesIsolation Thermique Hygrométrie HTMSebastien MENARDPas encore d'évaluation
- Devoir 01 2bacBIOF CorrigéeDocument8 pagesDevoir 01 2bacBIOF Corrigéewww.etudiantaymanemchibikPas encore d'évaluation
- Introduction À La RobotiqueDocument32 pagesIntroduction À La RobotiqueMehdi LaserPas encore d'évaluation
- Module Électricité 2007 1Document14 pagesModule Électricité 2007 1Serge Yannick MimboePas encore d'évaluation
- OAM Brief ReviewDocument5 pagesOAM Brief ReviewHøussàéîn Bèn MéssâõûdPas encore d'évaluation
- Carrelet TeDocument3 pagesCarrelet TeNeoXana01Pas encore d'évaluation
- Corrigé 2 TD EM SMP S4Document17 pagesCorrigé 2 TD EM SMP S4Khalid SalmiPas encore d'évaluation
- TP 02 Syst Lin Ass S4 Auto.1Document4 pagesTP 02 Syst Lin Ass S4 Auto.1moussaab yousfiPas encore d'évaluation
- Mise en Position Des PiècesDocument32 pagesMise en Position Des PiècesCker Jo100% (1)
- 2020-2021 - DS2 (Réponses)Document3 pages2020-2021 - DS2 (Réponses)mael.lemouroux2002Pas encore d'évaluation
- SUITE CHAPITRE I. Mur de SoutènementDocument9 pagesSUITE CHAPITRE I. Mur de SoutènementSafaa SassouPas encore d'évaluation
- Cours Conversion Dénergie Partie 1+partie2Document15 pagesCours Conversion Dénergie Partie 1+partie2JAMILA CIPas encore d'évaluation
- Stabilité Au Feu D'éléments de Structure Métallique Tendus Ou en Flexion Simple Sans Protection-2002Document9 pagesStabilité Au Feu D'éléments de Structure Métallique Tendus Ou en Flexion Simple Sans Protection-2002sautier_thomasPas encore d'évaluation
- Rev C1 4Sc PDFDocument2 pagesRev C1 4Sc PDFOmar MakhPas encore d'évaluation
- Equations Au Brevet-2Document5 pagesEquations Au Brevet-2vfpm88cxhsPas encore d'évaluation
- Informations Sur Les ManomètresDocument3 pagesInformations Sur Les Manomètresbayou100% (1)
- TD2Document1 pageTD2Yassin Si OmgharPas encore d'évaluation
- Assemblages Sous Chargement StatiqueDocument24 pagesAssemblages Sous Chargement Statiqueghasseme abdelbasitPas encore d'évaluation
- Chapitre 1 Les Éléments ChimiquesDocument7 pagesChapitre 1 Les Éléments Chimiquesoliviacali188Pas encore d'évaluation
- 8 Trace Crite Mesurer Des Longueurs 2 2Document7 pages8 Trace Crite Mesurer Des Longueurs 2 2Dav100% (1)
- Bac Blanc 2024 PC SERIE D DREN Béré SUJETDocument4 pagesBac Blanc 2024 PC SERIE D DREN Béré SUJETcoulibalyzakaria898Pas encore d'évaluation
- Théorie Des GroupesDocument14 pagesThéorie Des GroupesChantal MBIZIPas encore d'évaluation
- Présentation Duree de Vie de RoulementDocument69 pagesPrésentation Duree de Vie de Roulementkolandi coulibalyPas encore d'évaluation