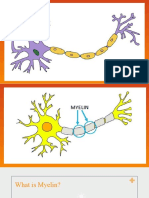
Dementia
Dementia
Download as pdf or txt
You might also like
- Genome Chapter SummariesDocument8 pagesGenome Chapter SummariesAshley YaoNo ratings yet
- EBOOK Infection Control and Management of Hazardous Materials For The Dental Team Download Full Chapter PDF KindleDocument62 pagesEBOOK Infection Control and Management of Hazardous Materials For The Dental Team Download Full Chapter PDF Kindlemichael.carathers448100% (50)
- MCAT CARS Practice Passages & ReviewDocument15 pagesMCAT CARS Practice Passages & ReviewSadek Mowakket100% (2)
- 20 Mechanisms of InjuryDocument2 pages20 Mechanisms of Injurypantufo100% (1)
- Alport SyndromeDocument49 pagesAlport SyndromemuslimNo ratings yet
- Nov 2017 Infantile Spasms WebinarDocument22 pagesNov 2017 Infantile Spasms WebinartetiNo ratings yet
- Ataxia TelangiectasiaDocument15 pagesAtaxia TelangiectasiaRose de DiosNo ratings yet
- Proteins As Products: Introduction To BiotechnologyDocument41 pagesProteins As Products: Introduction To BiotechnologyAulia Devi PurnamaNo ratings yet
- An Overview of DementiaDocument44 pagesAn Overview of DementiaAnil KakunjeNo ratings yet
- Neuro Degenerative DisordersDocument19 pagesNeuro Degenerative DisordersShalem. KNo ratings yet
- Pathophysiology of DementiaDocument3 pagesPathophysiology of DementiaIvy NNo ratings yet
- Disease of Nervous System Lect 2Document62 pagesDisease of Nervous System Lect 2Monirul IslamNo ratings yet
- AlcoholDocument20 pagesAlcoholrecklesspeshal2058100% (1)
- Vascular DementiaDocument3 pagesVascular DementiaAkhmad Ulil AlbabNo ratings yet
- STROKEDocument9 pagesSTROKEhillary elsaNo ratings yet
- Essential ThrombocytosisDocument12 pagesEssential ThrombocytosisGd Padmawijaya100% (1)
- Amyotrophic Lateral Sclerosis (ALS) : Group ThreeDocument20 pagesAmyotrophic Lateral Sclerosis (ALS) : Group ThreeosaeNo ratings yet
- Alzheimers-Disease-Genetics-Fact-Sheet 0Document8 pagesAlzheimers-Disease-Genetics-Fact-Sheet 0api-285676076No ratings yet
- Multiple Sclerosis. OCENA AUBREYDocument26 pagesMultiple Sclerosis. OCENA AUBREYCHRISTIAN RAY ALPAS PASILIAO100% (1)
- Basics of EpilepsyDocument15 pagesBasics of EpilepsyDiana CNo ratings yet
- Cerebrovascular Pathology: Abel B. (MD) Pathology Lectures, NMEI, DBUDocument59 pagesCerebrovascular Pathology: Abel B. (MD) Pathology Lectures, NMEI, DBUdenekeNo ratings yet
- Subacute Combined Degeneration of Spinal CordDocument4 pagesSubacute Combined Degeneration of Spinal CordPriyanka MathurNo ratings yet
- Chronic Kidney DiseaseDocument8 pagesChronic Kidney DiseaseAyiessa_AJNo ratings yet
- InhalDocument33 pagesInhallupeNo ratings yet
- Case 1 - Sickle Cell Disease - PPSXDocument44 pagesCase 1 - Sickle Cell Disease - PPSXNICHOLAS KAUMBA100% (1)
- Understanding Alzheimer DiseaseDocument35 pagesUnderstanding Alzheimer DiseasenadyaNo ratings yet
- Definition:: Between 16Document3 pagesDefinition:: Between 16NIDA MUSTAFANo ratings yet
- Multiple SclerosisDocument45 pagesMultiple Sclerosispriyanka bhowmikNo ratings yet
- Neurocutaneous SyndromesDocument8 pagesNeurocutaneous SyndromesvcNo ratings yet
- Delirium in Critically IllDocument37 pagesDelirium in Critically IllSanj.etcNo ratings yet
- Neuropathology: FK UisuDocument28 pagesNeuropathology: FK UisuAnggi WahyuNo ratings yet
- Delirious You or The Patient?Document34 pagesDelirious You or The Patient?Vishala MishraNo ratings yet
- Drugs For The Treatment of Diabetes MellitusDocument51 pagesDrugs For The Treatment of Diabetes MellitusRaga ManduaruNo ratings yet
- Vasculitic NeuropathiesDocument20 pagesVasculitic NeuropathiesHITIPHYSIONo ratings yet
- Causes of Stroke PDFDocument16 pagesCauses of Stroke PDFEmmanuel AguilarNo ratings yet
- Brain Tumor: Classification and External ResourcesDocument5 pagesBrain Tumor: Classification and External ResourcestheamaciasNo ratings yet
- EpilepsyDocument22 pagesEpilepsyRajeev RanjanNo ratings yet
- Hypnotics and Sedatives PDFDocument35 pagesHypnotics and Sedatives PDFSayan Nag100% (1)
- Classification of The Epilepsies: Purpose: For Clinical DiagnosisDocument25 pagesClassification of The Epilepsies: Purpose: For Clinical Diagnosisayu rifqiNo ratings yet
- Lipid Lowering DrugsDocument15 pagesLipid Lowering Drugsmwaithira71682No ratings yet
- Niemann Pick Type CDocument10 pagesNiemann Pick Type Capi-301744373No ratings yet
- Intracranial HypertensionDocument7 pagesIntracranial HypertensionIsabela OlgaNo ratings yet
- The Role of in The OF: CAM TreatmentDocument47 pagesThe Role of in The OF: CAM TreatmenterinahuntNo ratings yet
- Cognitive DisorderDocument72 pagesCognitive DisorderJamal P. AlawiyaNo ratings yet
- Epilepsy IndiaDocument72 pagesEpilepsy Indiaswathi bsNo ratings yet
- Identification and Diagnosis: Compulsive Alcoholic BeveragesDocument3 pagesIdentification and Diagnosis: Compulsive Alcoholic BeveragesKaren JulaoNo ratings yet
- Complications of Diabetes MellitusDocument46 pagesComplications of Diabetes MellitusAbhijith Menon100% (1)
- Types of DementiaDocument6 pagesTypes of DementiaDyah Ayu100% (1)
- Cerebellar Disorders: Dr. Mohamed Nasreldin HamdoonDocument14 pagesCerebellar Disorders: Dr. Mohamed Nasreldin HamdoonMohamed Nasreldin HamdoonNo ratings yet
- Head TraumaDocument15 pagesHead TraumaDede Yusuf FNo ratings yet
- Donepezil: DR Rashmi SurtiDocument55 pagesDonepezil: DR Rashmi SurtirsurtiNo ratings yet
- ENDOCRINOLOGYDocument63 pagesENDOCRINOLOGYYuni IndrianiNo ratings yet
- Sickle Cell Disease: Click To Edit Master Subtitle StyleDocument13 pagesSickle Cell Disease: Click To Edit Master Subtitle StyleAditya Rangga Fandiarta100% (1)
- Seizures and EpilepsyDocument76 pagesSeizures and EpilepsyManoj GhimireNo ratings yet
- Neurology Topics: Degenerative DiseasesDocument57 pagesNeurology Topics: Degenerative Diseasesलुकास विडालNo ratings yet
- Transient Ischemic Attack: - Time Based Definition: (Old Definition) Harrisons Principle of Medicine 19 EditionDocument14 pagesTransient Ischemic Attack: - Time Based Definition: (Old Definition) Harrisons Principle of Medicine 19 EditionHermie Alexander SiaNo ratings yet
- Approach To Pancytopenia: Moderator - DR Vishal Gupta MD Medicine Presented By-Dr Narendra Singh Resident Doctor 2Document35 pagesApproach To Pancytopenia: Moderator - DR Vishal Gupta MD Medicine Presented By-Dr Narendra Singh Resident Doctor 2ntnquynhproNo ratings yet
- Pathology of The Central Nervous System 2A2016Document63 pagesPathology of The Central Nervous System 2A2016Rose AnnNo ratings yet
- Diseases of ImmunityDocument55 pagesDiseases of ImmunityMeera ANN AJINo ratings yet
- Central Venous Catheter - StatPearls - NCBI BookshelfDocument11 pagesCentral Venous Catheter - StatPearls - NCBI Bookshelfsafrina100% (1)
- DementiaDocument9 pagesDementiaSivabharathi SivanandamNo ratings yet
- DementiaDocument38 pagesDementiarajikakurupNo ratings yet
- Seminar DementiaDocument63 pagesSeminar DementiaAhmad Syahmi YZNo ratings yet
- Dementia AssignmentDocument19 pagesDementia AssignmentVandna Vikram Novlani50% (2)
- DementiaDocument49 pagesDementiayussufNo ratings yet
- Central Nervous System Pathology: Presented byDocument70 pagesCentral Nervous System Pathology: Presented byNicholasNo ratings yet
- Chap. 4B The Three-Dimensional Structure of Proteins: TopicsDocument28 pagesChap. 4B The Three-Dimensional Structure of Proteins: Topicscatalina esanuNo ratings yet
- Neurocognitive DisordersDocument12 pagesNeurocognitive DisordersCYRELLE ANNE MALLARINo ratings yet
- Robbins Infectious DiseaseDocument27 pagesRobbins Infectious DiseaseJustine HungNo ratings yet
- Handbook of Clinical Neurology Human Prion Diseases 1St Edition Maurizio Pocchiari Full ChapterDocument67 pagesHandbook of Clinical Neurology Human Prion Diseases 1St Edition Maurizio Pocchiari Full Chapterjeremy.gibbons826100% (8)
- A Proposed Cleaning Classification System For Reusable Medical Devices To Complement The Spaulding ClassificationDocument11 pagesA Proposed Cleaning Classification System For Reusable Medical Devices To Complement The Spaulding Classificationmicrobehunter007No ratings yet
- Protein Aggregation Kinetics, Mechanism and Curve Fitting - A Review of The LiteratureDocument23 pagesProtein Aggregation Kinetics, Mechanism and Curve Fitting - A Review of The LiteratureravarNo ratings yet
- Mad Cow Dix Agarose LabDocument9 pagesMad Cow Dix Agarose LabZade DakkakNo ratings yet
- Protein AggregationDocument29 pagesProtein AggregationAndré PérezNo ratings yet
- Proceedings of The National Academy of Sciences Sackler NAS Colloquium Self-Perpetuating Structural States in Biology, Disease, and GenetDocument141 pagesProceedings of The National Academy of Sciences Sackler NAS Colloquium Self-Perpetuating Structural States in Biology, Disease, and GenetAndrei CorneaNo ratings yet
- BiochemDocument383 pagesBiochemtylermedNo ratings yet
- Scientific American - October 2014 USADocument104 pagesScientific American - October 2014 USAmarproofNo ratings yet
- MEDF1012A Amino Acids and ProteinsDocument48 pagesMEDF1012A Amino Acids and ProteinsminhyunxiiiiNo ratings yet
- Eugenics and AIDS Conspiracy Summary by DR Romesh Senewiratne-Alagaratnam MD (2012)Document12 pagesEugenics and AIDS Conspiracy Summary by DR Romesh Senewiratne-Alagaratnam MD (2012)Dr Romesh Arya ChakravartiNo ratings yet
- Creutzfeldt-Jakob Disease 1Document16 pagesCreutzfeldt-Jakob Disease 1anu1643No ratings yet
- Protein Structure and FunctionDocument35 pagesProtein Structure and FunctionPRAJWAL SHYAM BHOSALENo ratings yet
- Rt-Quic: A New Test For Sporadic CJD: Alison J E GreenDocument7 pagesRt-Quic: A New Test For Sporadic CJD: Alison J E GreenAliciaNo ratings yet
- Soils-The Good, The Bad, and The BeautifulDocument52 pagesSoils-The Good, The Bad, and The Beautifulkett8233No ratings yet
- Lab Medical International Vol.28.6-7Document68 pagesLab Medical International Vol.28.6-7Iguodala OwieNo ratings yet
- Virus, Viroids and PrionsDocument33 pagesVirus, Viroids and PrionsShivaveerakumar S. ChandrikimathNo ratings yet
- Classification of Infectious DiseaseDocument64 pagesClassification of Infectious DiseaseSaadTamNo ratings yet
- Jim Stone Freelance 4-13-2021Document95 pagesJim Stone Freelance 4-13-2021Brian CharlesNo ratings yet
- Chapter 04Document57 pagesChapter 04meena jeerhNo ratings yet
- MicrobiologyDocument28 pagesMicrobiologyMahrose NawazNo ratings yet
- Week 2 - Molecular BiologyDocument39 pagesWeek 2 - Molecular BiologyReginaldy FalNo ratings yet