Ferro Electric Materials
Ferro Electric Materials
Download as pdf or txt
You might also like
- Dielectric Properties of SolidsDocument40 pagesDielectric Properties of SolidsHannan MiahNo ratings yet
- Lattice VibrationsDocument10 pagesLattice VibrationsVarun GovindNo ratings yet
- Neutron DiffractionDocument30 pagesNeutron DiffractionJaba PriyaNo ratings yet
- Synchrotron NotesDocument10 pagesSynchrotron NotesjarjarbrightNo ratings yet
- Physics Lab ManualDocument55 pagesPhysics Lab Manualvncharymsc100% (1)
- Ferroelectric NotesDocument6 pagesFerroelectric Noteschvar80100% (1)
- Xxcaa (Rolf E. Hummel) Electronic Properties of MaterialDocument94 pagesXxcaa (Rolf E. Hummel) Electronic Properties of MaterialfonsekapdlNo ratings yet
- Magnetic MaterialsDocument38 pagesMagnetic MaterialsMuhammad Shahriar100% (1)
- 10 X-Ray DiffractionDocument8 pages10 X-Ray DiffractionProf.Dr.Mohamed Fahmy Mohamed Hussein100% (1)
- Unit 5 - Engg Physics NEP Superconductors and NanoDocument9 pagesUnit 5 - Engg Physics NEP Superconductors and NanojanhvilkwNo ratings yet
- Optical MicrosDocument15 pagesOptical MicrosDHASARAIAH SNEHA100% (1)
- Electrical Measurement Lab (EEE-352)Document23 pagesElectrical Measurement Lab (EEE-352)DhinakarrajNo ratings yet
- 1 Electromagnetic InductionDocument20 pages1 Electromagnetic InductionHarmanjeet SinghNo ratings yet
- Thermal Properties of SolidsDocument16 pagesThermal Properties of SolidsParie Perdana100% (1)
- Vicker Hardness TesterDocument4 pagesVicker Hardness TesterVijayakumar mNo ratings yet
- Rotational Raman Spectroscopy: The Polarizability of The Molecule Must Be AnisotropicDocument21 pagesRotational Raman Spectroscopy: The Polarizability of The Molecule Must Be AnisotropicKartik RanaNo ratings yet
- Diffraction PDFDocument72 pagesDiffraction PDFSuyash SarafNo ratings yet
- Many-Electron Atoms: Electron Spin Pauli Exclusion Principle Symmetric and Antisymmetric Wave FunctionsDocument33 pagesMany-Electron Atoms: Electron Spin Pauli Exclusion Principle Symmetric and Antisymmetric Wave FunctionssNo ratings yet
- Ferroelectricity: Spontaneous Dipole MomentDocument24 pagesFerroelectricity: Spontaneous Dipole Momentaliazab100% (2)
- General Physics Lab Report: Faculty of Engineering and TechnologyDocument19 pagesGeneral Physics Lab Report: Faculty of Engineering and TechnologyIzaNo ratings yet
- OscillationsDocument7 pagesOscillationsjayashriparida09No ratings yet
- Unit 4 Semiconductor Physics-Edited PDFDocument57 pagesUnit 4 Semiconductor Physics-Edited PDFMUSICAL MASTI RINGTONENo ratings yet
- Magnetic MaterialsDocument18 pagesMagnetic MaterialsAmitKumarNo ratings yet
- Density of States - Derivation PDFDocument5 pagesDensity of States - Derivation PDFsarathsrnairNo ratings yet
- Ferro Dia para Magnetism PDFDocument11 pagesFerro Dia para Magnetism PDFShanna-Kay Wood-Davidson100% (1)
- Superconductivity (New)Document9 pagesSuperconductivity (New)RshshNo ratings yet
- X-Ray DiffractionDocument30 pagesX-Ray DiffractionMerve Ayvaz KöroğluNo ratings yet
- Engineering Physics 2 Unit-3Document82 pagesEngineering Physics 2 Unit-3Sriram JNo ratings yet
- Dielectric Notes PDFDocument24 pagesDielectric Notes PDFSuresh VishnoiNo ratings yet
- Solid State Physics2 PDFDocument72 pagesSolid State Physics2 PDFTejasree KambamNo ratings yet
- Band Theory of SolidsDocument22 pagesBand Theory of Solidsmunku05100% (1)
- Lab Report #2 Bragg ScatteringDocument10 pagesLab Report #2 Bragg ScatteringJohn FiveNo ratings yet
- Chapter 2 Crystal StructureDocument38 pagesChapter 2 Crystal StructureAbo Abdo100% (1)
- Thin Film Deposition Technology: 1. Objective and Scope of This SectionDocument30 pagesThin Film Deposition Technology: 1. Objective and Scope of This SectionFast FeneNo ratings yet
- CHM361-CHAPTER 3 Crystalline & Solid StateDocument58 pagesCHM361-CHAPTER 3 Crystalline & Solid StateEhazNo ratings yet
- 6.2 Low Energy Electron Diffraction (LEED)Document9 pages6.2 Low Energy Electron Diffraction (LEED)Shams ShamsNo ratings yet
- MultiferroicsDocument21 pagesMultiferroicsprashantitbhushahi100% (3)
- Fermi EnergyDocument7 pagesFermi EnergyBobNo ratings yet
- Electrostatics PowerPointDocument37 pagesElectrostatics PowerPointJose GulitiwNo ratings yet
- Superconductivity, Superconducting Materials and AplicationsDocument8 pagesSuperconductivity, Superconducting Materials and Aplicationsgabriel_oltean09No ratings yet
- Wiedemann-Franz Law PDFDocument3 pagesWiedemann-Franz Law PDFArunraj KasiNo ratings yet
- Lecture 7 Application DTA & DSC01Document34 pagesLecture 7 Application DTA & DSC01ZUL KAMARUDDINNo ratings yet
- Ewald SphereDocument57 pagesEwald SphereMohammad Rameez0% (1)
- Band Theory of SolidsDocument12 pagesBand Theory of SolidsFitrianiNo ratings yet
- Magnetic PropertiesDocument20 pagesMagnetic Propertiespatrick saliwanNo ratings yet
- U1 MAgneticPropDocument19 pagesU1 MAgneticPropAbinash PandaNo ratings yet
- Experimental PhysicsDocument444 pagesExperimental PhysicsJerome MeccaNo ratings yet
- Free Electron TheoryDocument8 pagesFree Electron TheoryNeelam KapoorNo ratings yet
- Physical Metallurgy (Miller Indices)Document17 pagesPhysical Metallurgy (Miller Indices)Majid Ullah Sajid MahmoodNo ratings yet
- Lect 4 Ion ImplantationDocument28 pagesLect 4 Ion ImplantationPavankumar GnvaNo ratings yet
- DielectricsAndFerroelectricsDocument29 pagesDielectricsAndFerroelectricsDante Filho100% (1)
- Temperature and Heat: Heat Is A Flow of Energy Due To Temperature DifferencesDocument17 pagesTemperature and Heat: Heat Is A Flow of Energy Due To Temperature Differencespaulyn ramosNo ratings yet
- Scherrer Equation, Modified Scherrer Equation, Williamson-Hall PlotDocument8 pagesScherrer Equation, Modified Scherrer Equation, Williamson-Hall PlotJorge Luis Vazquez100% (1)
- Btech 1 Sem Engineering Physics Kas101t 2022Document2 pagesBtech 1 Sem Engineering Physics Kas101t 2022Ujjwal GargNo ratings yet
- Oscillations MCQ Simple Harmonic Motion: Question H1: Why Study This Stuff?Document8 pagesOscillations MCQ Simple Harmonic Motion: Question H1: Why Study This Stuff?hhhhhNo ratings yet
- London EquationsDocument5 pagesLondon Equationsyehtt0212No ratings yet
- Introduction To NanotechnologyDocument18 pagesIntroduction To NanotechnologyTushar PanditNo ratings yet
- Dieletric ppt-1Document18 pagesDieletric ppt-1YASH JAINNo ratings yet
- Semiconductor Physics - UG 1Document43 pagesSemiconductor Physics - UG 1aranyachakraborty7No ratings yet
- Module 4 2021-22Document15 pagesModule 4 2021-22Rohith ReddyNo ratings yet
- Convocation Application FormDocument1 pageConvocation Application FormMohanrajRajangamNo ratings yet
- Modeling of An Automotive Exhaust Thermoelectric Generator: This Is A Dummy TextDocument163 pagesModeling of An Automotive Exhaust Thermoelectric Generator: This Is A Dummy TextMohanrajRajangamNo ratings yet
- Tva Bok 0008504 Naadi VaagadamDocument458 pagesTva Bok 0008504 Naadi VaagadamMohanrajRajangamNo ratings yet
- Material's Science & TechnologyDocument38 pagesMaterial's Science & TechnologyMohanrajRajangamNo ratings yet
- Non-Destructive Testing (NDT)Document52 pagesNon-Destructive Testing (NDT)MohanrajRajangamNo ratings yet
- Characteristics: ProcessingDocument3 pagesCharacteristics: ProcessingMohanrajRajangamNo ratings yet
- A 'Photo' That Won't Fit in The Family Album: Engineers Create 3D Model of Baby's Face For Partially-Sighted MotherDocument1 pageA 'Photo' That Won't Fit in The Family Album: Engineers Create 3D Model of Baby's Face For Partially-Sighted MotherMohanrajRajangamNo ratings yet
- The Use of 3D Scanning in The Turbine Blade Industry.....Document1 pageThe Use of 3D Scanning in The Turbine Blade Industry.....MohanrajRajangamNo ratings yet
- Click To Edit Master Title StyleDocument5 pagesClick To Edit Master Title StyleMohanrajRajangamNo ratings yet
- 3D Scanners UK Scan & Create Polygon Ammonite ModelDocument1 page3D Scanners UK Scan & Create Polygon Ammonite ModelMohanrajRajangamNo ratings yet
- 731A - P31 Operation ManualDocument7 pages731A - P31 Operation ManualAlvaro CotaquispeNo ratings yet
- W63AHxNB VFBGA178 PKG Datasheet A01-005 20200728Document122 pagesW63AHxNB VFBGA178 PKG Datasheet A01-005 20200728Sudhir SainiNo ratings yet
- How Fiber Optic Cables WorkDocument13 pagesHow Fiber Optic Cables WorkMuhammad Sharif JanjuaNo ratings yet
- Introduction and NoiseDocument65 pagesIntroduction and NoiseBAHARUDIN BURAHNo ratings yet
- Toshiba PDFDocument2 pagesToshiba PDFsygabrielaNo ratings yet
- Light FixtureDocument5 pagesLight FixturedesignNo ratings yet
- Nortel Optical Metro - Carritech TelecommunicationsDocument2 pagesNortel Optical Metro - Carritech TelecommunicationsCarritech TelecommunicationsNo ratings yet
- INVENTORY OF AV-EQUIPMENT SY 2000-2016finalDocument15 pagesINVENTORY OF AV-EQUIPMENT SY 2000-2016finalRomeo Dela CruzNo ratings yet
- P 31 NMR Spectroscopy 2Document11 pagesP 31 NMR Spectroscopy 2김동완No ratings yet
- KZGDocument2 pagesKZGAdilson BogadoNo ratings yet
- GT-1000-JMTCD (H2) Portable Hydrogen Gas Detector SpecificateDocument4 pagesGT-1000-JMTCD (H2) Portable Hydrogen Gas Detector SpecificateSigid AriewibowoNo ratings yet
- Uba 2071Document35 pagesUba 2071Floricica Victor VasileNo ratings yet
- Datasheet Hybrid H T1 Series Global EN - 1023 - Web 6Document3 pagesDatasheet Hybrid H T1 Series Global EN - 1023 - Web 6Ionut Robert BalasoiuNo ratings yet
- PNP Transistor OperationDocument5 pagesPNP Transistor OperationmohanNo ratings yet
- Motorola PTP 300 Series User GuideDocument288 pagesMotorola PTP 300 Series User GuideMauricio SurcoNo ratings yet
- 2004 05 Product Catalog Eltek PDFDocument72 pages2004 05 Product Catalog Eltek PDFwolfdeniroNo ratings yet
- OmniScan MX2 Getting Started Manual (2011)Document2 pagesOmniScan MX2 Getting Started Manual (2011)Raul MedinaNo ratings yet
- DT V24G1 020510us PDFDocument2 pagesDT V24G1 020510us PDFgibonulNo ratings yet
- AUO T240HW01 V0 DatasheetDocument3 pagesAUO T240HW01 V0 DatasheethandokoNo ratings yet
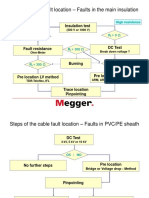
- Cable Fault Location ProcedureDocument12 pagesCable Fault Location ProcedureAshari Endra100% (1)
- 22-Range Auto Pocket Size Digital Multimeter: Taking MeasurementsDocument2 pages22-Range Auto Pocket Size Digital Multimeter: Taking MeasurementsGustavo RodriguezNo ratings yet
- Manual Pedalera ZOOM 4040Document29 pagesManual Pedalera ZOOM 4040Oskar Nuñez BeltranNo ratings yet
- Apr-Pc QuestDocument99 pagesApr-Pc Questridip2009No ratings yet
- Radio Propagation and Propagation Path Loss ModelsDocument42 pagesRadio Propagation and Propagation Path Loss ModelsNoshin FaiyroozNo ratings yet
- Digital. PPT - Chapter 4Document40 pagesDigital. PPT - Chapter 4JenberNo ratings yet
- En - CBT-N (1) - CBT 60NDocument7 pagesEn - CBT-N (1) - CBT 60NboroumandNo ratings yet
- PSoC Assembly Language User GuideDocument110 pagesPSoC Assembly Language User GuidekmmankadNo ratings yet
- Feedback Feedback Topologies PDFDocument12 pagesFeedback Feedback Topologies PDFOrlando Jose Heredia0% (1)
- Parallel OperationDocument4 pagesParallel Operationashad ashaNo ratings yet
- CNCDocument89 pagesCNCpepeNo ratings yet