

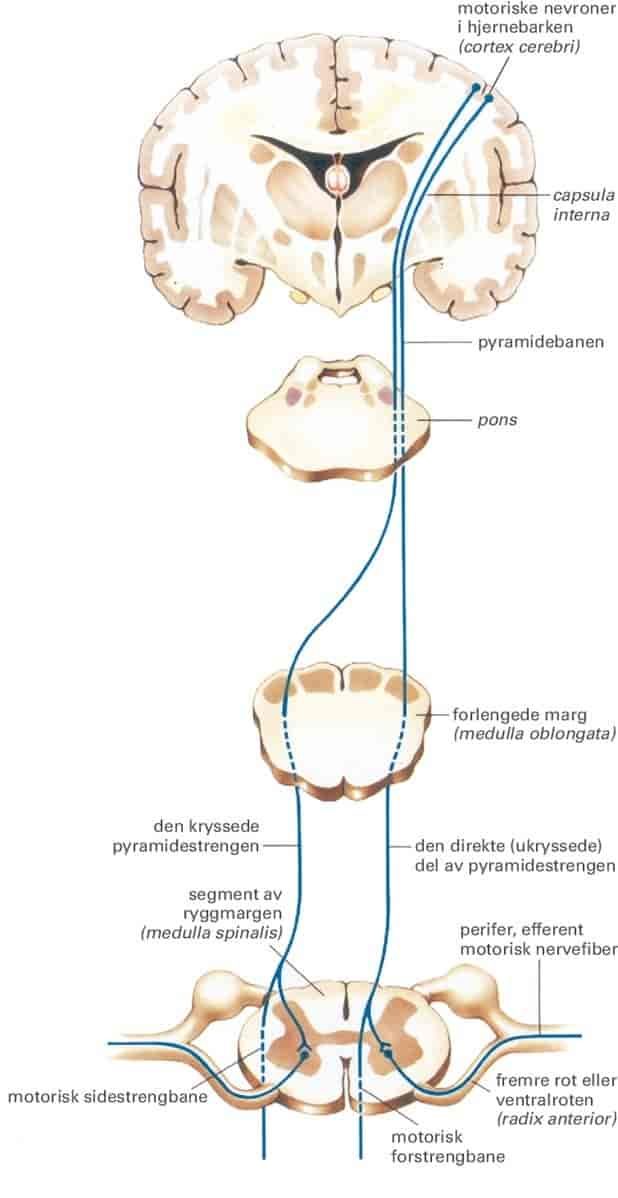
ALS er en sykdom som rammer bestemte nerveceller i hjernen og ryggmargen. Det er bare nerveceller som er med på å styre skjelettmuskulaturen som rammes. Noen av de vanlige tegnene på ALS er unormale reflekser, muskelsvakhet og lammelser. Andre deler av nervesystemet rammes ikke. Det betyr at man fortsatt kan se, høre og forstå språk. Hukommelsen er bevart.
Sykdommen er en nevrodegenerativ sykdom, det vil si at nervecellene som rammes, dør. Årsaken til sykdommen er ukjent. ALS er den vanligste formen for motornevronsykdom.
Sykdommen utvikler seg med ulik hastighet fra person til person, men i mange tilfeller medfører sykdommen utbredte lammelser og død i løpet av noen få år. ALS diagnostiseres først og fremst gjennom en klinisk undersøkelse, men undersøkelsesmetoder som for eksempel elektromyografi og MR-undersøkelse brukes også.


Kommentarer
Kommentarer til artikkelen blir synlig for alle. Ikke skriv inn sensitive opplysninger, for eksempel helseopplysninger. Fagansvarlig eller redaktør svarer når de kan. Det kan ta tid før du får svar.
Du må være logget inn for å kommentere.